命名
早在1887年,Minkowski 记录了垂体肿瘤(pituitary tumor)病例,并命名为“acromegaly”(肢端肥大症),在1915年,就有学者提到了“adenoma of pituitary gland” ,在1932年,Summers 就用了“pituitary adenoma”进行病例报道。截止2023年仍在使用垂体腺瘤(pituitary adenoma)来称呼这一疾病。但在2017年,在“14th Meeting of the International Pituitary Pathology Club, Annecy, France”(第14届国际垂体病理俱乐部会议)上,与会专家指出因垂体腺瘤不具备典型的良性肿瘤特征且部分病例具有侵犯性和侵袭性,并有致死风险,不再适合用一个用于良性肿瘤的称呼 “adenoma”,鉴于腺垂体细胞具有神经内分泌细胞特征,建议用“pituitary neuroendocrine tumor(pitNET)”(垂体神经内分泌瘤)来命名该疾病。在2022年出版的“世界卫生组织(WHO)神经内分泌和内分泌肿瘤分类(第5版)”中正式引入了“pituitary neuroendocrine tumor(pitNET)”(垂体神经内分泌瘤)。
分类
垂体腺瘤有多种分类标准,常用的有依据瘤体大小分类和依据瘤体内分泌功能分类,此外还有依据组织病理染色表现的分类。
依据瘤体大小分类:
微腺瘤:指瘤体直径<1cm者;
大腺瘤:指瘤体直径>1cm者,瘤体直径>3cm者可称为巨大腺瘤。
依据肿瘤细胞组织学和功能分类:
指应用免疫组织化学方法检测瘤体内特定激素水平,结合患者临床表现和血清激素含量进行的分类。目前多采用世界卫生组织(WHO)2017年垂体瘤病理分类,其按激素免疫组化、细胞分化来源谱系进行分类。
泌乳素细胞腺瘤(PRL 瘤)
占垂体腺瘤的40%~60%,男性表现为阳痿、性功能减退等,女性表现为闭经-泌乳综合征。患者血液泌乳素水平明显升高,病理检查多为嫌色细胞;免疫组化检测PRL阳性,垂体特异POU 类同源结构域转录因子1(PIT-1)阳性,雌激素受体α(ERα)阳性 。
生长激素细胞腺瘤(GH 瘤)
占有功能腺瘤的20%~30%,儿童期发病表现为巨人症,成年期发病表现为肢端肥大症,患者血浆GH升高,病理组织染色呈嗜酸性细胞为主,免疫组化检测GH阳性,PIT-1阳性,部分可有PRL阳性及ERα阳性。
促肾上腺皮质激素细胞腺瘤(ACTH 瘤)
占垂体腺瘤的5%~15%,临床表现为皮质醇增多症(Cushing 综合征,库欣综合征),患者血液ACTH水平升高导致全身脂肪、蛋白质代谢紊乱。病理检查嗜碱性细胞或嫌色细胞为主,免疫组化检测ACTH阳性, 低分子量细胞角蛋白(LMWCK)阳性,T-box 家族成员TBX19(T-PIT)阳性 。
促甲状腺激素细胞腺瘤(TSH 瘤)
罕见,不足1%,临床表现为甲亢或甲减,患者血浆TSH升高,病理检查免疫组织化学染色βTSH阳性,α-亚基阳性,PIT-1阳性。
促性腺激素细胞腺瘤(gonadotroph adenoma,LH/FSH 瘤)
占无功能性垂体腺瘤的70%~90%,多为大腺瘤,临床以肿瘤占位效应为主要表现,内分泌功能异常者少见。病理检查免疫组织化学染色β-FSH 和β-LH阳性,α-亚基(不同组合)阳性, 类固醇生成因子1(SF-1)阳性, 锌指转录调控蛋白的GATA 家族成员(GATA-2)阳性,ERα阳性。
多分泌功能细胞腺瘤
由2种或2种以上的内分泌细胞肿瘤组成,分为PIT-1阳性多激素腺瘤和不常见的免疫组化组合腺瘤,临床表现为多种内分泌功能失调。前者免疫组化GH、PRL、β-TSH和PIT-1均阳性,部分α亚基可阳性,后者有ACTH+GH型、ACTH+PRL型,免疫组化可见相应激素阳性,还有其他转录因子阳性。
零细胞腺瘤(null-cell adenoma)/无内分泌功能细胞腺瘤(non-functioning adenomas)
既往报道该类型占25%~35%,主要指无明显内分泌功能异常的垂体腺瘤,以上各个类型腺瘤均可出现,但相对少见,临床表现以垂体占位效应为主,已有报道70%~90%的该类型腺瘤为促性激素细胞腺瘤。病理检查细胞分化程度低,无明显分泌功能,免疫组化检测各个激素及转录生长因子均为阴性。
依据肿瘤生长特性分类
世界卫生组织(WHO)2017年垂体瘤分类中还有根据肿瘤的生长特性的分类法,将垂体瘤分为典型垂体腺瘤、高危型垂体腺瘤和垂体癌。高危型垂体腺瘤包括稀疏颗粒型生长激素腺瘤、男性催乳素腺瘤、沉默型促肾上腺皮质激素腺瘤、Crooke细胞腺瘤和PIT-1 阳性多激素腺瘤。当肿瘤发生转移诊断为垂体癌。
WHO 垂体神经内分泌瘤分类(2022年)
该分类系统依据肿瘤细胞谱系、细胞类型及相关特征进行分类。
PIT-1-谱系垂体神经内分泌瘤
生长激素瘤
泌乳素瘤
泌乳素-生长激素细胞瘤
促甲状腺激素瘤
成熟型多激素性 PIT-1谱系瘤
未成熟型 PIT-1谱系瘤
嗜酸性干细胞瘤
生长激素和泌乳素混合型瘤
TPIT-谱系垂体神经内分泌瘤
SF1-谱系垂体神经内分泌瘤
细胞谱系未明型垂体神经内分泌瘤
依据病例组织染色表现分类:
腺瘤组织在常规苏木精-伊红(HE)染色后,显微镜下观察可分为:嗜酸性细胞(约占垂体前叶总细胞数的35%)、嗜碱性细胞(约15%)和嫌色性细胞(约50%),依据瘤体细胞表现,可分为嗜酸性细胞腺瘤、嗜碱性细胞腺瘤、嫌色性细胞腺瘤和混合性细胞瘤。
病因和发病机制
垂体腺瘤的确切病因尚不十分清楚,发病机制也有待进一步研究,可能与下列因素有关:
激素分泌失常
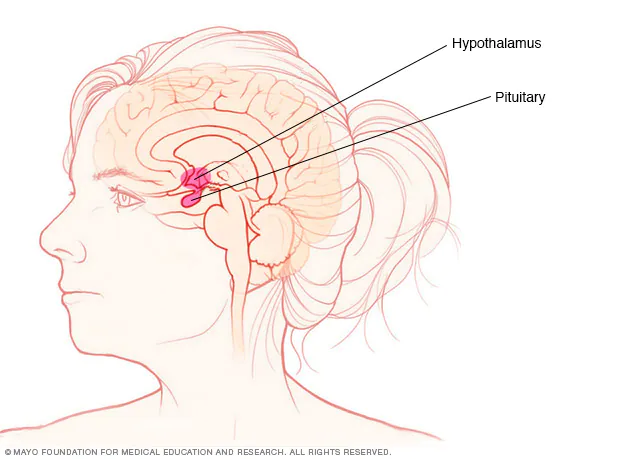
下丘脑的促释放激素和垂体内自有激素的旁分泌或自分泌作用可能在垂体瘤形成的促进阶段起一定作用。PRL 瘤,GH 瘤,LH/FSL 瘤,TSH 瘤、ACTH 瘤与相应的促释放激素分泌增多或者下丘脑释放抑制因子减少有关。某些生长因子,如PTH相关肽( PTHrP) 、血小极衍化生长因子( PDGF ) 、转化生长因子α和β(TGFα 和TGFß ) 、白介素(IL)、胰岛素样生长因子1(IGF-1)等在不同垂体瘤中都有较高水平的表达,它们可能以旁分泌或自分泌的方式促进垂体瘤细胞的生长和分化。靶激素分泌减少(如甲状腺激素和皮质醇)是促发功能性垂体瘤的重要原因,如慢性原发性甲状腺功能减退症患者可发生垂体TSH 瘤。
无功能垂体瘤虽然不能在血循环中检测到激素水平升高,但免疫组化和分子生物学研究发现肿瘤细胞能产生糖蛋白激素(LH、FSH、TSH)的 α 和 β 亚单位,可能由于翻译后修饰缺陷,导致不能产生具有生物活性的激素。
肿瘤相关基因表达变化
细胞周期调控异常,涉及cyclin、CDK、CDK 抑制因子、Rb 和E2F的复杂调控;
染色体稳定性异常,如垂体肿瘤转化基因(PTTG)的高表达;
信号通路异常,如GNAS 突变导致的Gsα持续激活、Akt、Wnt 信号通路异常;
旁分泌的生长因子和细胞因子异常,涉及FGF 、EGF , VEGF 、NGF 等;
抑癌转录因子失活或表达减少,如MEG3 、PLAGL1、GADD45γ;
促肿瘤转录因子增加,如HMGA2。
基因突变
主要与家族遗传性垂体瘤有关,约5% 的垂体瘤为家族遗传性,大约5 % 的垂体瘤为家族性遗传综合征所导致,最常见的包括多发性内分泌腺瘤病1 型(MEN- 1 )、4 型( MEN -4) , Carney 综合征,家族性孤立性垂体腺瘤,分别由MEN- l 、p27、PRKARIA ,AIP 基因突变导致。在散发性垂体瘤中,基因突变少见, 目前仅GNAS 、PIK3AC , USP8 和USP48 分别在部分生长激素腺瘤、各类型垂体瘤和ACTH 腺瘤中发现存在体细胞突变。
表观遗传调控异常
表观遗传调控异常可能参与垂体瘤的发生,如MEG3 、PLAGL1 、GADD45-γ 被发现启动子甲基化而沉默;组蛋白H3K9乙酰化增加, 一定程度上与p53 错误表达相关。
流行病学
因为部分垂体腺瘤体积过小或无内分泌功能而不能被临床发现,故垂体腺瘤具体的患病率尚不能明确,一般以临床诊断病例为依据。一项来自芬兰北部的群体研究结果显示垂体腺瘤的标准化发病率约4.0/10万人,其中泌乳素瘤为2.2/10万人,临床无功能腺瘤为1.0/10万人,生长激素腺瘤为0.34/10万人,促肾上腺皮质激素腺瘤为0.17/10万人。来自英国超过8万人的一项群体研究结果显示所有垂体腺瘤的标准化患病率为77.6/10万人,其中泌乳素瘤为44.4/10万人,无功能腺瘤为22.2/10万人,生长激素腺瘤为8.6/10万人,肾上腺皮质激素腺瘤为1.2/10万人。一项回顾性研究(1969年~1993年6月)显示,接受手术治疗的2230例垂体瘤患者,泌乳素瘤、ACTH瘤和TSH瘤的女性患者多于男性,而无功能腺瘤和生长激素瘤则是男性多于女性;泌乳素瘤多见于20~50岁者,无功能腺瘤大部分在40~80岁。来自马耳他共和国的流行病学调查显示,垂体瘤患者首次诊断平均年龄为40.6 (SD ±15.0)岁,年龄在30~39岁者标准化发病率最高,为 7.42/100,000人/年。
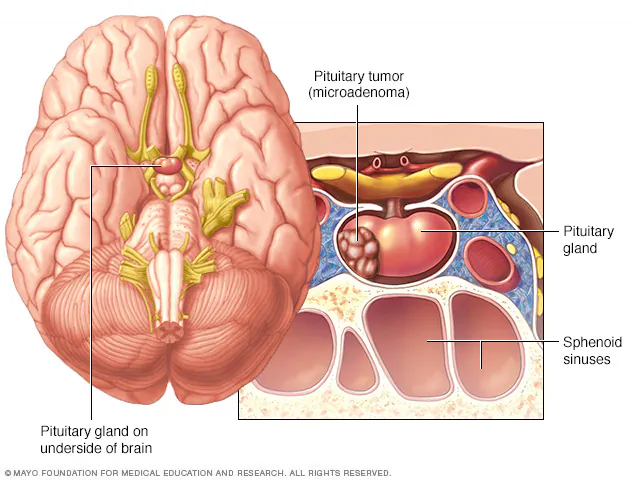
病理
90% 的垂体瘤为良性腺瘤,少数为增生,极少数为腺癌。多数为单个,小肿瘤生长于蝶鞍内,多呈球形或卵圆形,表面光滑;大者往往向蝶鞍外发展而呈不规则结节状,有包膜,可侵蚀和压迫视交叉、下丘脑、第三脑室和附近的脑组织。应用HE 染色中腺垂体细胞分为嗜酸性、嗜碱性、嫌色性,其中嫌色性细胞腺瘤占80% ,已有研究证明大部分嫌色细胞具有激素分泌功能,如GH 腺瘤、ACTH 腺瘤、PRL腮瘤等均为嫌色细胞腺瘤。结合患者临床表现用垂体激素抗体做免疫组化可鉴定肿瘤种类,但阴性不能排除诊断,因为细胞合成的激素可不形成颗粒直接分泌入血。电镜下可观察到不同功能肿瘤其激素分泌颗粒致密度和大小有区别,PRL 瘤和 GH 瘤颗粒较大而 ACTH 瘤颗粒较小。但是病理表现和临床激素检测有时不完全一致,因此诊断必须结合组织形态、免疫组化和临床表现后决定。
临床表现
有功能垂体腺瘤在发病早期可出现相应激素分泌亢进症状,而无功能腺瘤无明显激素功能异常症状,后者多在颅脑CT或MRI检查时发现,称意外瘤(accidentenoma);当瘤体增大使正常腺体组织受压,可能会出现垂体其他激素分泌不足导致相应激素功能低下症状,当垂体瘤过大还会压迫周围组织而产生相应症状。故垂体腺瘤的临床表现包括肿瘤导致的占位侵袭和激素异常分泌症候群。若垂体癌发生颅外转移,可产生相应的临床表现,较为罕见。
腺垂体受压症候群
由于肿瘤压迫正常垂体组织,垂体促激素分泌减少,导致相应靶腺萎缩,激素分泌减少。由于腺垂体代偿能力很强,受压破坏60% 才开始出现轻微症状,破坏95%有严重功能减退。腺垂体受压破坏时最先受影响的是促性腺激素细胞和生长激素细胞,随后出现促甲状腺激素分泌减少,促肾上腺皮质激素不足一般发生在后期。因此促性腺激素不足症状最常见,妇女表现为闭经、不育,男性可出现性欲减退、阳痿等。由于垂体门脉系统受压致PRL分泌抑制因子(PIF,即多巴胶)减少,60%~80% 患者出现 PRL 分泌增多。
垂体周围组织受压症候群
垂体腺瘤增大压迫周围组织可出现相应症状,症状取决于肿物解剖位置、大小和扩展方向。骨性鞍区背侧鞍顶是压力最薄弱处,因此垂体瘤常侵犯鞍上区域。
头痛:早期约2/3 病人有头痛,主要位于眶后,前额和双颠部,程度轻,间歇性发作,多系肿瘤直接剌激或鞍内压增高,引起垂体硬膜囊及鞍隔受压所致。当肿瘤突破鞍脯,鞍内压降低, 疼痛则可减轻或消失。晚期头痛可因肿瘤向鞍旁发展侵及颅底硬膜及血管和压迫三叉神经而引起。少数巨大腺瘤鞍上发展突入第三脑室,造成室间孔或导水管梗阻,出现颅内压增高时头痛较剧。或肿瘤坏死、出血,瘤内压力急剧增高。如瘤壁破裂致垂体卒中性蛛网膜下腔出血者为突发剧烈头痛,并伴其他神经系统症状。
视神经结构受累:60%~80%患者因肿瘤向鞍上侵犯压迫视交叉,出现不同程度视功能障碍,典型者为双颞侧偏盲。早期可表现为双颞侧上限视野缺损,然后逐步扩大到双颞侧下象限,进而鼻侧视野受累,最终可以导致失明。另外,视神经受到侵犯或脑脊液回流障碍也会导致视力减退。长期视交叉受压会导致视神经萎缩。
垂体卒中:垂体腺瘤有时可因出血、梗死而发生垂体卒中。起病急骤,表现为额部或一侧眶后剧痛,可放射至面部,井迅速出现不同程度的视力减退,严重者可在数小时内双目失明,常伴眼球外肌麻,尤以第 Ⅲ 对脑神经受累最为多见,也可累及第 Ⅳ、VI 对脑神经,并可出现神志模糊、定向力障碍、颈项强直甚至昏迷。部分患者发生急性肾上腺皮质功能衰竭,可的现血压下降,低钠血症,低血糖症等。
其他部位受累:肿瘤向侧方侵袭累及海绵窦,可造成第 Ⅲ、Ⅳ、VI 对脑神经及第 V 对脑神经的眼支及上颌支麻痹,患者可出现不同程度复视、上险下垂、面部感觉减退等;垂体肿瘤侵犯鞍底可使蝶窦受累。若侵袭性肿瘤侵犯颚顶,可引起鼻咽部的梗阻、感染或脑脊液漏,但此情况较少发生。罕见颞叶和额叶受累,患者可出现沟回癫痫、人格障碍或嗅觉缺失。侵袭性垂体肿瘤直接侵犯下丘脑可能导致重要的代谢异常,包括体温异常、食欲异常、睡眠障碍、中枢性尿崩症、口渴、性早熟或性腺功能减退等。
有功能腺瘤激素分泌异常症候群
有激素分泌功能的垂体腺瘤根据其分泌激素不同,表现出相应的激素功能亢进症状。
PRL 瘤:患者泌乳素增高、雌激素减少,女性表现为闭经、溢乳、不孕,又称为Forbis-Albright 综合征;男性表现为性欲减退、阳痿、不育。
GH 瘤:可分泌过多GH,引起骨骼、软组织、脏器过度生长,发病在青春期前骨骺未融合者表现为巨人症( gigantism) ,发生在青春期后,骨骺已融合者则表现为肢端肥大症(acromegaly) 。
垂体ACTH 瘤,腺瘤持续分泌过多ACTH 使肾上腺皮质增生进而导致分泌过多皮质醇,导致库欣病(Cushing’s Syndrome)。
TSH 瘤:罕见,由于 TSH 分泌过多,血T3、T4水平增高,同时可导致甲状腺肿大,出现甲状腺功能亢进症状。
LH/FSH 瘤:有人认为罕见,但也有报道有70%~90%的无功能腺瘤实质为促性腺激素细胞腺瘤,早期无明显症状,晚期有性功能减退、闭经、阳痿、睾丸萎缩等表现。
诊断
垂体腺瘤诊断分3个层次:①垂体瘤的确定;②明确垂体瘤类型和性质;③了解垂体功能及其周围组织受累情况。对于有临床表现的垂体腺瘤,依据患者症状、体征及实验室内分泌学检查和影像学检查不难作出诊断。但对于早期微腺瘤,因为无明显临床症状或体征,在内分泌学检查和影像学检查也中无明显异常,其诊断存在很大困难。
实验室检查
主要检测腺垂体激素分泌情况。无功能腺瘤可能导致垂体功能减退,如生长激素缺乏、促性腺激素缺乏等,同时可能导致PRL 水平升高。功能性垂体腺瘤根据其来源不同应行相应的激素检查。当怀疑垂体腺瘤时,初步的激素检查应包括:① 血清PRL; ② GH、胰岛素样生长因子-1 (IGF-1);③ ACTH、血皮质醇;④ FSH、LH、雌二醇、孕酮、睾酮;⑤ TSH、FT3、FT4。
影像学检查
核磁共振成像(MRI):是垂体肿瘤首选影像诊断方法。在T1加权显像上,垂体腺瘤较周围正常组织信号低,而在T2 加权像上信号加强。同时还可以评估瘤体位置、大小、与周围组织关系及受累情况。较大肿瘤中出现低信号区提示坏死或囊性变,出现高信号区提示出出血。垂体不对称或垂体柄分离是垂体腺瘤的间接征象。
计算机断层扫描(CT):可见垂体占位病变密度高于脑组织,增强后可提高肿瘤检出率,较 MRI 能更好地显示瘤体周围骨质情况,并有助于与空泡蝶鞍鉴别。
眼科检查
主要为视觉及视野评估,有助于判断视交叉受累情况及程度。
鉴别诊断
垂体腺瘤为应和以下疾病相鉴别。
颅咽管瘤:颅咽管瘤常与垂体腺瘤相混,多发生在鞍内,常向第三脑室内、鞍后或鞍旁发展。典型颅咽管瘤不难鉴别,多发生在儿童或青春前期,表现为垂体内分泌功能低下,发育停滞, 50% 呈株儒型或矮小症。约1/3 病人患有尿崩症。蝶鞍可正常或扩大,有时后床突破坏,附近骨质侵蚀, 70% 的病人鞍上或(和)鞍内呈现钙化斑块,肿瘤多呈囊性,有时囊壁钙化呈特有的蛋壳形。CT 扫描为鞍上低密度囊性区。边界清楚、圆形、卵圆形或分叶状,实体肿瘤CT扫描表现为均匀的密度增高区,囊壁呈壳样钙化是颅咽管瘤的特点,有助于诊断和鉴别珍断。注射造影剂,实体肿瘤为均匀增强;囊性肿瘤为环形囊壁增强。MRI 显示鞍上、鞍内的囊性肿物,可为长T1、T2,也可为短T1、T2 信号。手术时见肿瘤内为绿色液体,有时囊液稠如机油,内含胆固醇结晶。在成人,颅咽管瘤多为实质性,可有视力视野障碍,内分泌功能减退等,难与垂体腺瘤鉴别,有时取下瘤组织作病理检查,才能确定诊断。
Rathke 囊肿,为一种先天发育异常,患者通常没有症状,部分患者依囊肿位置及大小不同可出现不同程度的头痛及视力障碍,女性患者可出现闭经。垂体功能减退及脑水肿较少见。CT检测占位病变多低密度;MRI 检测病变液体信号明显,增强扫描无强化。
空泡蝶鞍综合征,分为先天性和继发性两类。先天性者系鞍膈先天性缺损或形成不全(占21.5% ) ,68%~87%为中年经产妇,与妊娠分娩的生理性垂体体积增大有关。继发性者为垂体手术和放射线疗法后所致。一般无症状,CT 扫描为蝶鞍内的低密度区,诊断关键为脑池造影CT 扫描,发现造影剂(METRlZAMIDE) 进入蝶鞍的蛛网膜下腔。如有脑脊液漏及进行性视力视野障碍是手术适应证。
垂体炎,可分为4种类型,即淋巴细胞性垂体炎(最常见)、肉芽肿性垂体炎、IgG4垂体炎、黄瘤病性垂体炎和坏死性垂体炎。。垂体炎与垂体瘤最常用的鉴别手段是MR1。在MRI上,淋巴细胞性垂体炎表现为垂体对称性增大、呈三角形或哑铃形、平扫条件下等信号、增强后均匀强化,垂体柄增粗居中,伴尿崩症的患者神经垂体高信号消失。黄瘤病性垂体炎常表现为囊性、环状强化的病灶。肉芽肿性垂体炎的影像学表现仍未阐明,而坏死性垂体炎MR1表现不特异。FDG-PET 可用于提示垂体炎症性疾病,特别是IgG4垂体炎和朗格汉斯细胞组织细胞增生症。
治疗
垂体瘤的治疗目标是缓解局部压迫、维持正常垂体激素水平、保护正常垂体细胞功能、防止腺瘤复发。目前垂体瘤的治疗方法包括手术、放疗和药物治疗。应根据肿瘤性质、大小、局部压迫等情况综合判断选择合适的治疗方案。
手术治疗
除催乳素瘤外,手术治疗是垂体瘤的首选治疗方式。经蝶手术是目前垂体瘤手术最常用术式,但明显向鞍上和侧向侵犯的垂体肿瘤,经蝶手术无法完全去除肿物,需行开颅手术治疗。术后肿瘤未能完全切除者需辅以放疗或药物治疗,伴垂体功能低下者尚需激素替代治疗。垂体腺瘤手术治疗后死亡发生率约0.27%~2%。
经鼻蝶入路手术:适用于: ①存在症状的垂体腺瘤卒中; ②垂体腺瘤的占位效应引起压迫症状; ③难以耐受药物不良反应或对药物治疗产生抵抗的催乳素腺瘤及其他高分泌功能的垂体腺瘤(主要为 ACTH 瘤、GH 瘤); ④垂体部分切除和(或)病变活体组织检查术; ⑤经鼻蝶手术的选择还需考虑到以下几个因素:瘤体的高度、病变形状、瘤体的质地与血供情况、鞍隔面是否光滑完整、颅内及海绵窦侵袭的范围大小、鼻窦发育与鼻腔病理情况、患者全身状况及手术意愿。
开颅垂体腺瘤切除手术:适用于不能行经蝶窦入路手术者;鼻腔感染患者。
联合入路手术:主要针对肿瘤主体位于鞍内、鞍上、鞍旁发展,呈“哑铃”形者。
放射治疗
主要作为手术和内科药物治疗的辅助手段,可分为外照射和内照射。外照射是国内常用的方法,包括高能射线治疗、重粒子放射治疗、立体定向放射治疗(γ 刀)等。内照射即通过开颅手术(额路)或经鼻腔穿过蝶窦途径将放射性物质植入蝶鞍当中进行放射,其放射源包括放射性核素钇-90( Y) 、金-198( Au) 等。近年来高能射线发展,己取代了常规X 线治疗。放射治疗指征包括存在手术禁忌,手术无法完全切除,术后复发但病灶不大,且不宜再行手术者。目前国内使用最多的是立体定向放射治疗,其并发症主要包括腺垂体功能减退、继发脑瘤、脑血管病、视力损伤、脑坏死。
药物治疗
药物治疗可分为针对有功能腺瘤的治疗和激素不足的激素替代治疗。根据垂体腺瘤类型,选用不同的药物治疗方案。
有功能腺瘤治疗:
多巴胶受体激动剂主要用于 PRL 瘤的治疗,该类药物可使 PRL 水平迅速下降,并可缩小肿瘤体积,还可用于 GH 瘤的治疗。常用药物有澳隐亭、卡麦角林等。
生长抑素类似物可抑制多种激素分泌,如 GH 和 TSH 等,可用于治疗肢端肥大症和 TSH 分泌型肿瘤。常用生长抑素类似物有奥曲肤、兰瑞肤等。
GH 受体拮抗剂(培维索孟)可阻断 GH 生物学作用,也可用于肢端肥大症的治疗。
抑制类固醇生物合成的药物可于库欣病的辅助治疗,如酮康唑、甲吡酮、米托坦等。米非司酮可拮抗皮质醇作用,也可用于Cushing 综合征的治疗。赛庚啶与血清素的竞争剂,可抑制血清素的作用,用于ACTH 瘤的辅助治疗。
激素替代治疗
适用于垂体功能不足患者,可因垂体腺瘤压迫引起,也可为腺瘤术后并发症,依据病情,可予相应激素治疗。
预后
由于多数垂体瘤是良性肿瘤,经蝶显微外科手术切除垂体腺瘤手术治愈率约60%~90%。垂体微腺瘤易于完全切除,GH微腺瘤治愈缓解率达57%~90%;PRL 微腺瘤在33%~90%;ACTH 微腺瘤达74%~90%;大腺瘤疗效一般在30%~70%。对侵蚀性大腺瘤则很难彻底切除,只能改善症状,难以根治,患者常需终身随访及治疗。垂体瘤手术前视力受损严重者,术后恢复的可能性较小。无功能腺瘤的临床转归一般较好。垂体癌预后不佳。
垂体瘤术后复发率约7%~35%,单纯肿瘤切除者复发率可达50%,PRL 腺瘤5 年复发率可达40%,ACTH 腺瘤复发率为10% 。