克拉霉素(Clarithromycin)属于半合成的大环内酯类抗生素,可与细菌核糖体50S亚基结合,从而抑制其蛋白合成而产生抗菌作用,可用于敏感菌引起的咽炎、扁桃体炎、中耳炎、鼻窦炎、支气管炎、肺炎、皮肤感染,还可与其他药物联用治疗幽门螺杆菌感染。
本页面主要目录有关于克拉霉素的:医学用途、药理机制、药代动力学、风险与禁忌、风险提示、历史、使用情况、化学信息、专利等介绍

克拉霉素(Clarithromycin)属于半合成的大环内酯类抗生素,可与细菌核糖体50S亚基结合,从而抑制其蛋白合成而产生抗菌作用,可用于敏感菌引起的咽炎、扁桃体炎、中耳炎、鼻窦炎、支气管炎、肺炎、皮肤感染,还可与其他药物联用治疗幽门螺杆菌感染。
本页面主要目录有关于克拉霉素的:医学用途、药理机制、药代动力学、风险与禁忌、风险提示、历史、使用情况、化学信息、专利等介绍
克拉霉素
Clarithromycin
克拉仙、卡斯迈欣、百红优
片剂、胶囊剂、干混悬剂、颗粒剂、缓释片
适用于敏感菌所致上呼吸道感染、下呼吸道感染以及皮肤和软组织感染
口服、静脉滴注
化学药
乙类
处方药
克拉霉素常见不良反应有味觉障碍、腹痛、腹泻、恶心、呕吐、消化不良等胃肠道反应以及头痛;严重者可出现重症多形性红斑、中毒性表皮剥脱性坏死、严重过敏反应、肝毒性、肝功能衰竭或艰难梭菌引起的假膜性肠炎。
对大环内酯类药物过敏者、存在水电解质紊乱者、孕妇及哺乳期妇女、伴肾功能损害的重度肝功能损害者以及患有室性心律失常或有QT间期延长等疾病者禁用。
克拉霉素部分制剂已纳入医保,其中克拉霉素片、克拉霉素胶囊为医保乙类药品;克拉霉素干混悬剂、克拉霉素缓释片未纳入医保。较红霉素,克拉霉素的抗菌活性更强。
克拉霉素适用于对其敏感的致病菌引起的感染,包括:
化脓性链球菌引起的咽炎和扁桃体炎;
流感嗜血杆菌、卡他莫拉菌及肺炎链球菌所致急性鼻窦炎、儿童中耳炎;
流感嗜血杆菌、副流感嗜血杆菌、卡他莫拉菌及肺炎链球菌所致慢性支气管炎急性细菌感染性加重;
流感嗜血杆菌、肺炎链球菌、肺炎支原体或肺炎衣原体所致肺炎;
敏感金黄色葡萄球菌或化脓性链球菌所致单纯性皮肤及软组织感染;
鸟分枝杆菌或胞内分枝杆菌感染的预防与治疗;
与其他药物联合用于幽门螺杆菌感染的治疗。
牙源性感染的治疗。
(1)成人:口服,一次250~500mg,一日2次,疗程7~14日;静脉滴注,一次500mg,一日2次,疗程一般7~14日。
(2)6个月以上的儿童:①每次7.5mg/kg,一日2次口服。②或按以下方法口服给药:体重8~11kg者,每次62.5mg,一日2次;体重12~19kg者,每次125mg,一日2次:体重20~29kg者,每次187.5mg,一日2次;体重30~40kg者,每次250mg,一日2次。
(3)肾功能严重减退成人患者:可按下表调整用量。
肾功能严重减退患者的克拉霉素用量调整肌酐清除率>30 ml/min(0.5 ml/s) | 肌酐清除率<30 ml/min(0.5 ml/s) |
500mg,一日2次 250mg,一日2次 | 首剂500mg,以后每次250mg,一日2次 每次250mg,一日1次 |
(4)与其他抗菌药联合治疗幽门螺杆菌感染:一次0.5g,一日2次,餐后口服,疗程7日或10日(对于耐药严重的地区,可考虑适当延长至14日,但不宜超过14日)。
名称 | 规格 | 剂型 | 甲乙类 |
克拉霉素片 | 0.05g;0.125g;0.25g | 口服常释制剂 | 乙类 |
克拉霉素胶囊 | 0.125g;0.15g;0.25g | 口服常释制剂 | |
克拉霉素干混悬剂 | 0.125g | 口服混悬剂 | |
克拉霉素颗粒 | 0.05g;0.1g;0.125g;0.25g | 颗粒剂 | 乙类 |
克拉霉素缓释片 | 0.5g | 缓释控释剂型 | |
参考资料: | |||
克拉霉素属于半合成的大环内酯类抗生素,可与细菌核糖体50S亚基结合,从而抑制其蛋白合成,对多种需氧和厌氧的革兰阳性和革兰阴性菌均具有抗菌作用。对甲氧西林敏感葡萄球菌属和链球菌属的抗菌作用较红霉素略强。其体内代谢产物14-羟克拉霉素与克拉霉素对流感嗜血杆菌具有协同抗菌作用,较红霉素强2~4倍。对嗜肺军团菌、沙眼衣原体及溶脲脲原体的作用较红霉素为强。对幽门螺杆菌亦具有良好抗菌作用。对鸟分枝杆菌及龟分枝杆菌有抑制作用,对麻风分枝杆菌亦有抗菌作用。
克拉霉素对胃酸较稳定,口服后生物利用度为55%。单次口服400mg后tmax为2.7小时,Cmax为2.2mg/L;每12小时口服250mg后的稳态血药浓度约为1mg/L。克拉霉素及其主要代谢产物在体内分布广泛,鼻黏膜、扁桃体及肺组织中的药物浓度较同期血药浓度为高,血浆蛋白结合率65%~75%。
克拉霉素在肝脏中广泛代谢,代谢产物主要通过胆汁从粪便排泄;10%~15%以代谢产物从尿排泄。单次给药后t1/2为4.4小时,每12小时口服250mg和500mg后t1/2分别为3~4小时和5~7小时。低剂量给药(每12小时250mg)后经粪、尿两类途径排出的药量相仿,尿排出量约为给药量的32%;但剂量增大(每12小时500mg)时,尿中排出量较多。克拉霉素的药动学特点呈非线性动力学,随剂量而改变,口服高剂量后由于代谢饱和,母药的峰浓度超比例增加。
主要有味觉障碍、腹痛、腹泻、恶心、呕吐、消化不良等胃肠道反应以及头痛。严重的有:重症多形性红斑、中毒性表皮剥脱性坏死、严重过敏反应、肝毒性、肝功能衰竭或艰难梭菌引起的假膜性肠炎。静脉注射可引起静脉炎。
如果摄入过高剂量的克拉霉素,可能会出现胃肠道的不良反应。一位患有双向情感障碍的患者摄入8克克拉霉素后出现精神状态改变、偏执、低血钾和低氧血症。一旦发现克拉霉素服药过量,应立即去除尚未吸收的药物,并开展相应的支持治疗。与其他大环内酯类药物相似,克拉霉素的血清浓度不会被血液透析或腹膜透析影响。
克拉霉素禁止与阿司咪唑、西沙必利、匹莫齐特、特非那定合用,可能导致QT间期延长和心律失常(包括室性心律失常、室颤和尖端扭转型室速)。
禁止与麦角胺或双氢麦角胺合用,可能导致麦角碱中毒。
克拉霉素不应与HMG-CoA还原酶抑制剂(他汀类药物,如洛伐他汀或辛伐他汀)合用,否则可能增加横纹肌溶解的发生风险。
与洛美他派合用可能导致转氨酶显著升高,不得将克拉霉素与洛美他派联用。
与卡马西平、利福平、苯妥英等合用时,可能影响卡马西平的代谢,需监测卡马西平的血药浓度。
克拉霉素禁止与口服咪达唑仑、秋水仙碱、替卡格雷或雷诺嗪合用,可能增加不良反应的发生。
与利托那韦合用,可明显抑制克拉霉素的代谢,导致血药浓度增加。故二者合用时,应注意避免克拉霉素的每日剂量超过1000mg。
肌酐清除率<25ml/min或有急性血卟啉症者,不推荐联合使用克拉霉素与雷尼替丁和枸橼酸铋。
合用地高辛与克拉霉素时,应注意密切监测患者血清地高辛浓度。
HIV感染的成年人同时口服克拉霉素和齐多夫定时,后者的药物吸收会受影响,使其稳态血浓度下降,因此应错开服用时间(至少间隔4小时)。
应谨慎合用克拉霉素与经CYP3A4代谢的钙通道阻滞剂(如维拉帕米、氨氯地平、地尔硫卓),可能增加低血压发生风险。
克拉霉素可与不经CYP3A4代谢的药物(如苯妥英钠和丙戊酸钠)发生相互作用,建议合用时进行血药浓度监测。
孕妇及哺乳期妇女用药:孕妇和哺乳期妇女服用克拉霉素的安全性尚未确认,克拉霉素可由乳汁排出。孕早期和孕中期克拉霉素暴露会增加流产风险。故在没有风险/效益评估时,禁用于孕妇及哺乳期妇女。
儿童用药:6个月以上儿童可使用克拉霉素混悬液,6个月以下幼儿的安全性和有效性尚未确定,慎用任何剂型。
老年用药:65岁以上老年人慎用克拉霉素。
孕妇及哺乳期妇女禁用。
对大环内酯类药物过敏者禁用。
肝功能损害、中重度肾功能损害患者慎用。
存在水电解质紊乱者(低钾血症或低镁血症患者,有延长QT间期的风险)禁用。
某些心脏病(包括心律失常、心动过缓、Q-T间期延长、缺血性心脏病、充血性心力衰竭等)患者禁用。
美国食品和药物管理局(FDA)警告,使用克拉霉素会增加心脏问题或死亡的风险,并建议心脏病患者考虑替代抗生素。CLARICOR试验观察到,使用克拉霉素治疗2周后,冠心病人的死亡率出现了升高。
克拉霉素又名甲红霉素、克红霉素,是红霉素的衍生物。1952年,美国礼来公司开发出第一个大环内酯类抗生素——红霉素。为了改善红霉素的活性谱、给药频率和耐受性,1980年,日本大正制药公司的Wantanabe在红霉素结构中6位上由甲氧基取代羟基得到克拉霉素,1982年大正制药申请的专利中,Watanabe申请专利采用的路线是, 2′-O, 3′-N-二 (苄氧羰基) -N-去甲基红霉素A→甲基化→还原甲基化→克拉霉素。
1985年,大正制药与美国雅培公司合作获得国际版权,雅培公司也于1991年10月获得美国食品药品监督管理局对克拉霉素的批准。1990年,克拉霉素在爱尔兰、意大利上市。1991年6月,大正制药将其名为Clarith的品牌药物推向日本市场。1993年,以Klacid在中国香港上市。克拉霉素于2004年在欧洲成为仿制药,并于2005年年中在美国成为仿制药。
克拉霉素还可用于生物被膜病、抗肿瘤、血小板减少症、肝病、不稳定型心绞痛等。
在中国,克拉霉素是国家基本药物。
化学名称:6-O-甲基红霉素
化学结构式:
分子式:C38H69NO13
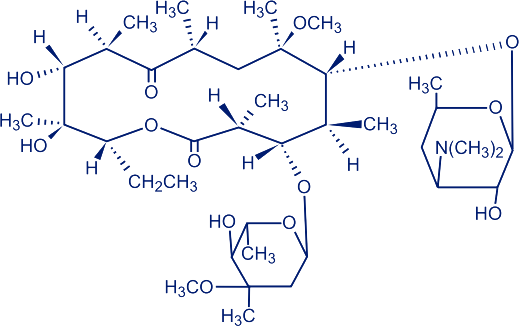
分子量:747.96
性状:白色或类白色结晶性粉末,无臭
水溶性:在丙酮或乙酸乙酯中溶解,在甲醇或乙醇中微溶,在水中不溶
密度:1.2±0.1g/cm
沸点:805.5±65.0 °C
熔点:217-220°C
日本大正制药为克拉霉素专利持有人,于1980年在日本申请专利,1981年在美国申请专利。1985年,大正制药将国际专利授予雅培,日本、韩国、台湾除外。克拉霉素日本专利分别于2000年6月4日和2004年9月1日到期。2004年11月19日,克拉霉素专利在英国和荷兰到期;20日,在德国、奥地利和比利时到期。2005年5月,在美国到期;10月4日,在瑞士和列支敦士登到期。2006年5月27日,在瑞典到期。2008年5月7日,在法国到期。

新亚洲娱乐(英文名:ASIA ENTERTAINMENT GROUP LIMITED,全称:新亚洲娱乐联盟集团有限公司)是一家以从事戏剧制作人及杂项戏剧服务为主的企业,成立于1999年,位于香港特别行政区。旗下分公司包括虎威艺能创作有限公司(TGS HK)、稻草人娱乐创作社(Scarecrow Entertainment)、虎威王朝音乐创作股份有限公司(TGS Music)、虎威活力娱乐传播有限公司(TGS Taiwan)、AK Entertainment(Korea)以及AEG Korea等。
印度孟买SENSEX30指数(又称孟买敏感指数)为印度最被广泛使用的指数,为投资印度的重要参考指标,是由孟买证券交易所发行。由于各类媒体提到的“印度股市”,实际上都是孟买股票交易所,因此,该交易所的SENSEX-30指数几乎成了印度股市的代名词。




