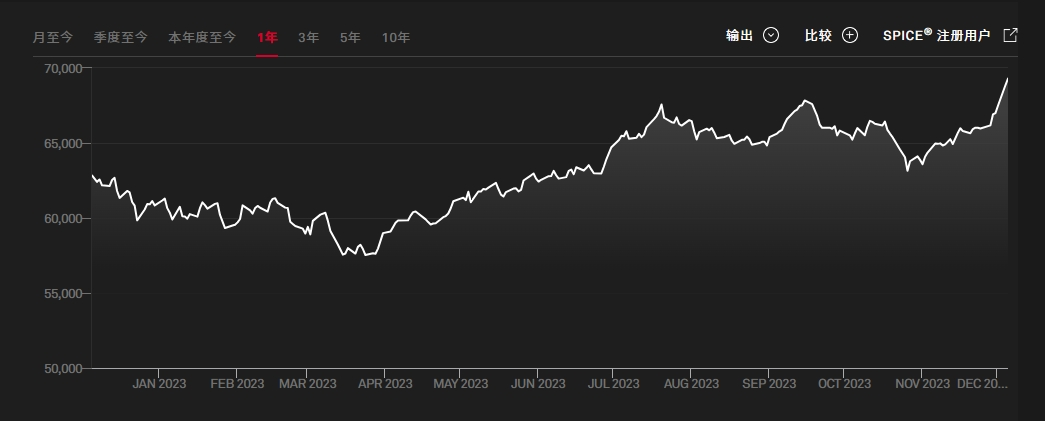
病因
尼曼—匹克病是由基因突变引起的,由于细胞溶酶体中缺乏神经鞘磷脂酶(sphingomyelinase,SMase),使得神经鞘磷脂不能水解而沉积于单核巨噬细胞和神经组织细胞中所致。神经鞘磷脂酶广泛存在于肝、脾、肾、脑和小肠等器官中。当其活力降低至正常50%以下,就不能降解代谢产生的神经鞘磷脂,导致神经鞘磷脂不同程度地沉积于全身各脏器:
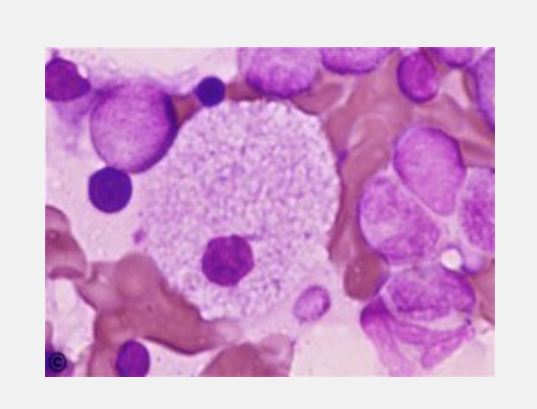
1、当神经鞘磷脂蓄积在单核巨噬细胞中,可出现肝、脾和淋巴结肿大;
2、当其沉积在中枢神经系统细胞中,中枢神经系统发生退行性病变;
3、当其沉积于视网膜,表现为眼底樱桃红斑,严重者失明;
4、当其累积在肺部,肺泡弥漫性受累,可影响呼吸功能;
5、当其沉积到骨骼,可引起骨质疏松,骨髓腔扩大,骨皮质变薄;
6、当其蓄积在肠壁神经丛细胞中,可出现腹泻。
根据基因突变可分为两种:
1、SMPD1基因突变导致酸性鞘磷脂酶缺陷,鞘磷脂广泛蓄积在内脏及中枢神经系统,导致尼曼-匹克氏病,表现为A型、B型及中间型。
2、NPC1、NPC2基因突变,表现为C型。
NPC1或NPC2基因突变导致胆固醇转运及吞噬障碍,引起细胞内未酯化的胆固醇蓄积和鞘脂代谢障碍。
临床分类
1、根据基因突变可分为四类:
(1)SMPD1基因突变引起的A型:临床表现严重,早期即可出现中枢神经系统退行性变。
(2)SMPD1基因突变引起的B型:多为慢性进展,内脏受累严重,较少累及中枢神经系统。
(3)SMPD1基因突变引起的,临床表现介于A型、B型之间的中间型。
(4)NPC1、NPC2基因突变引起的C型:发病年龄范围比较广,从围产期至成人期都可能发病。
2、根据临床表现分类
(1)急性神经型:多在出生后3~6个月发病,病情发展较迅速。
(2)非神经型:在婴幼儿或儿童期发病,病程进展缓慢。
(3)幼年型:儿童期发病,首次发病表现为肝脾肿大。
(4)Nova-Scotia型:临床经过较幼年型缓慢。
(5)成年型:成人发病,智力正常,无神经系统症状,不同程度肝脾肿大,可长期生存。
症状
尼曼—匹克病多见于婴幼儿,也有发生在新生儿时期。主要症状为肝脾肿大、贫血,病程较长者会出现营养不良、发育迟缓。
具体症状可分为五种类型:
1、A型
A型为典型的尼曼匹克病,大多出生后3~6个月内发病。他们在宫内及出生表现为正常。有的在新生儿期会有黄疸迁延,这种发生的几率比较小。出生后数周内出现因肌力、肌张力低下而发生喂养困难,体重难以增加,常伴有反复呕吐、腹泻等;3~6个月时出现肝脾增大和淋巴结肿大,病情发展迅速,6个月时即可出现精神运动发育衰退征象,表情淡漠、动作发育迟缓、听视力逐渐丧失、惊厥发作等,皮肤有棕黄色素沉着,约半数患儿可见眼底黄斑部樱红斑。多数会因为感染在4岁前死亡。
2、B型
患者发病较A型稍晚,多数在婴幼儿或儿童期发病,病程进展慢,大多以肝脾肿大、肝肝增大为主要特点,无神经系统症状,肝功能受损情况亦少见,患者多身材矮小,肺部因弥漫性浸润而易发生感染。一般不影响生命。
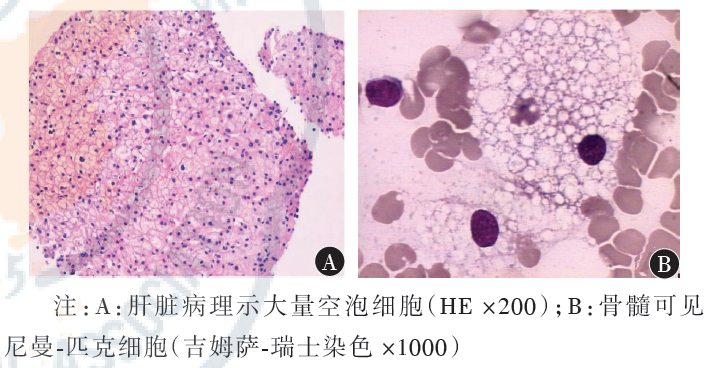
3、C型
多见儿童期发病,主要症状包括脾脏进行性肿大、肌张力增高、共济失调、精神分裂症样症状及认知功能减退等,出生后第一天或前几周即出现新生儿胆汁郁积性黄疸,50%患儿伴有进行性肝脾肿大。在婴幼儿早期多数只出现肝脾肿大症状。2岁以后逐渐出现神经系统症状,表现为出肝脾肿大,语言落后、智力运动障碍等。少年期表现为脾肿大,注意力低下、学习困难,,患儿行为笨拙、吞咽困难。青少年期一些患者除了脾肿大,还会精神异常、共济失调。
4、D型
具有加拿大Nova Scotia血统的特有患者类型,与C型有类似的临床表现,发病比C型缓慢,有明显黄疸、肝脾肿大和神经症状,多数在学龄期死亡。
5、E型
较为少见,症状轻微,有不同程度肝脾肿大,可长期生存。
诊断
诊断原则
根据尼曼-匹克氏病典型的临床表现,再结合血常规、血生化检查、酶学检查、骨髓穿刺检查、肝、脾和淋巴结活检、X线检查等相关辅助检查做出诊断。在诊断过程中,还需排除戈谢病、糖原积病Ⅰ型(GSDI)、高雪氏病婴儿型、Wolman病、神经节苷酶脂病I型、Hurler病等疾病。
诊断依据
1、临床症状
肝脾巨大、身体发育障碍、肌张力减退和中枢神经系统退行性病变,部分患者的皮肤、黏膜可见黄染,眼底可见樱桃红斑。
2、辅助检查
(1)骨髓检查、外周血中淋巴细胞和单核细胞有空泡对于该病诊断至关重要。
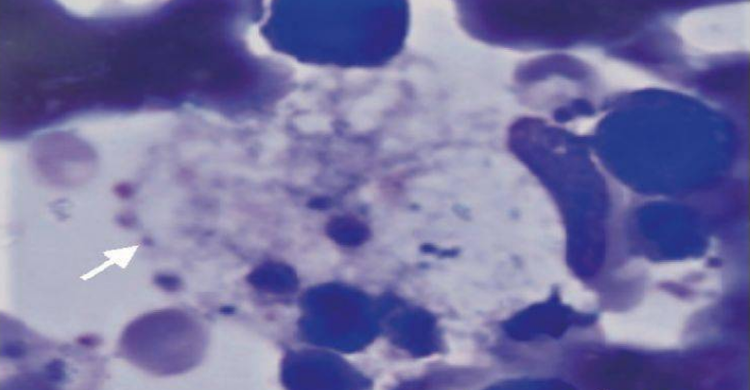
(2)血生化检查、肝、脾和淋巴结活检、X线检查、基因检测分析等相关辅助检查对该病的评估有一定的意义。
鉴别诊断
1、尼曼—匹克病与戈谢病的鉴别
尼曼儿克病与戈谢病的临床表现非常类似,尤其在以肝大脾大为主要表现时,在临床上难以区分。
这两种病的鉴别诊断,主要通过测定相应的葡萄糖脑在脂酶活性,或对戈谢病的致病基因 GBA进行测序。
2、尼曼—匹克病与糖原积病Ⅰ型(GSDI)的鉴别
尼曼—匹克病B型需与糖原累积病作区分,糖原积病Ⅰ型(GSDI)的主要特征是糖原和脂肪堆积在肝脏、肾脏.引起肝增大和肾增大。尼曼—匹克病B型是神经鞘磷脂酶的缺乏引起肝脾肿大。糖原积病Ⅰ型(GSDI)的确诊,通过测定患者的葡糖-6-磷酸酶或葡萄糖-6-磷酸移位酶活性,或对患者的G6PC或SLC37A1基因进行检测分析
3、尼曼—匹克病与Wolman病的鉴别
两者均会出现肝脾肿大、神经系统功能障碍的症状。但Wolman病患者无眼底樱桃红斑,X线腹部平片可见双肾上腺肿大,外形不变,有弥漫性点状钙化阴影。尼曼-匹克氏病患者肾上腺一般无异常。
4、尼曼—匹克病与Hurler病的鉴别
两者均会出现肝脾大、智力差、淋巴细胞浆有空泡,骨髓有泡沫细胞等。但Hurler病患者心脏缺损,多发性骨发育不全,无肺浸润。尿黏多糖排出增多,中性粒细胞有特殊颗粒,尼曼-匹克病则没有这种症状。
检查
1、实验室检查:血常规,查看血红蛋白正常或具有轻度贫血,脾亢时显时白细胞减少。血生化检查看肝功是否异常、血脂代谢是否异常。酶学检查,看磷脂酶的活性是否异常。
2、影像学检查:X线检查:长期存活患者可有骨质疏松、髓腔变宽、皮质变薄、长骨局灶性破坏;婴儿期以后肺泡受脂肪组织细胞浸润,肺部可见类似组织细胞增生X症的表现,但都无特异性。
3、病理学检查:肝、脾或淋巴结活检,取少量病变组织进行活检。骨髓穿刺,查看是否含有典型的尼曼-匹克细胞。
4、基因检测:看是否有SMPD1、NPC1、NPC2基因。
治疗护理
治疗
尼曼-匹克氏病尚无有效治疗方案,针对患者出现的类型不同,给予不同的治疗方案。对诊断为尼曼-匹克病患儿,应及时进行系统检查和随访观察。
1、对于血小板计数减少的患儿,出血重时给予输血等,对发生症状的肺浸润患儿,可视情况给子吸氧,或支气管肺灌洗术等专科处理。
2、药物治疗:该患者无特效药物,临床一般予抗氧化剂进行治疗,相关药品有:苯巴比妥、卡马西平、地西泮、新斯的明、维生素C、维生素E、丁羟基二苯乙烯,可以阻止鞘磷脂M所含不饱和脂肪酸的过氧化和聚合作用。
3、手术治疗:造血多功能干细胞移植,大多数尼曼-匹克病 B型患儿的肝功能损害较常见,必要时可进行肝移植;脾切除:适用于非神经型有脾功能亢进者,用于巨脾产生的机械症状。
康复护理
针对患儿的功能水平给予相应的训练,增强功能水平,防止继发性损伤,从而提高患儿的生活质量。
1、给予患儿唇周及口腔周围肌群良性刺激,帮助患儿提高吸吮能力和咀嚼能力。
2、家长每日强化和重复单个字的简单指令,增强患儿对词汇的理解,激发他们表达的欲望。
3、通过对关节的良性挤压手法增强本体感觉的输入,帮助患儿更好地维持功能位置和进一步活动。
4、针对累及肺脏或卧床为主的患儿,对患儿进行缩唇呼吸、腹式呼吸的训练,帮助他们增加气道阻力以及提高肺通气功能。
5、通过运动功能干预,提高患者的活动能力,防止肌肉萎缩。
饮食
科学合理的饮食习惯对患者恢复有一定帮助:
1、饮食营养要均衡,适当搭配蛋白、脂肪和碳水化合物。
2、适量进食新鲜蔬菜、水果补充维生素。
3、愉快地进餐对稳定病情,病情恢复有一定作用。
饮食禁忌
1、避免辛辣刺激性食物,如辣椒、芥末、姜、蒜等,防止加重脾胃负担。
2、成年患者应戒酒,酒精可损伤消化系统,引起疾病加重。
预防
1、对于有家族遗传史的患者,要做遗传咨询;
2、产前检查,一般在孕中期行羊水穿刺或四维彩超。
就诊科室
儿科、神经内科、血液内科。