简介 该病临床上多表现为进行性呼吸困难伴有刺激性干咳,双肺可闻及Velcro啰音,常有杵状指(趾)。胸部X线片主要表现为双肺底和周边分布的弥漫性网格状、蜂窝状阴影,肺功能为限制性通气障碍。病情一般进行性发展,最终因呼吸衰竭导致死亡。
IPF的直接致病因子尚不清楚,即病因不明,但该病的发生与一些危险因素有关,如遗传因素、吸烟、环境暴露、病毒感染及胃食管返流等。
IPF的治疗仍然是临床医师面临的难题。除肺移植能延长严重IPF生存期及改善患者的生活质量外,现有的药物,如吡非尼酮和尼达尼布能延缓肺功能下降速度,但不能阻止病情进展。需通过氧疗,肺康复等支持和对症处理改善患者生活质量;关注IPF急性加重、胃食管返流、肺动脉高压等常见并发症评价和处理。
IPF确切的患病率和发病率尚不清楚。IPF的患病率及发病率远比以前估计得高,同时死亡率也呈上升趋势。
病因 危险因素 IPF的直接致病因子尚不清楚,因此被冠以“特发性”,即病因不明。但诸多证据表明该病的发生与一些危险因素有关。
遗传因素:以下事实提示遗传因素或先天性易感因子可能与该病的发病有关:①家族性肺纤维化的病例在世界各地均有报道,且数量不断增加,这种病例多见于嫡亲和单卵双胞胎;②某些已知遗传疾病患者的肺纤维化发病率很高;③同样暴露于已知可引起肺纤维化的环境中,但仅有少数发病;④动物实验发现,特定的鼠系对发生肺纤维化有遗传易感性。
吸烟:虽然约 1/3的IPF 发生在终生不吸烟者,但多数的临床研究证实吸烟增加 IPF 发生的危险性,其暴露程度与IPF的发生率呈正相关,尤其是吸烟大于400支/年者。
环境暴露:暴露于某些金属粉尘(黄铜、铅及钢铁)和木质粉尘(松木)者的患病风险显著增加。其他粉尘暴露,如理发业、鸟类饲养、石材切割和抛光等也可能与IPF的发生有关。IPF患者尸体解剖发现肺部淋巴结内可厂无机物颗粒,也支持 IPF环境学病因。
病毒感染:某些病毒在 IPF发生中是否发挥重要作用一直受到学者们的关注。支持病毒感染与IPF 发病机制之间存在联系的主要证据是流行病学研究结果。高达97%的 IPF 患者肺中可以检测到EB病毒、巨细胞病毒、丙型肝炎病毒和人疱疹病毒中的一种或多种。因此推测,慢性病毒感染作为一种免疫刺激剂,引起慢性增殖性或炎性环境,导致肺纤维化的发生。但也有不支持这一观点的流行病学资料。关于病毒感染的病因假说仍存在不少争议。
胃食管返流:动物实验和临床研究均发现长期反复的胃内容物吸入可导致肺纤维化的发生,因此胃食管反流(GER)与IPF的关系受到重视。也有人认为,IPF患者减低的肺顺应性导致胸膜腔压力在吸气时较正常人更低,导致食管和食管下段括约肌功能不全,故而发生了GER,即其可能是IPF的结果,而非病因。
发病机制 致病因素导致肺泡上皮损伤和上皮下基底膜破坏,致使胶原纤维和细胞外基质过度生成。损伤的肺泡上皮和炎症细胞浸润的白细胞分泌TNF-α、TGF-β和IL-8等,诱导固有成纤维细胞增生,趋化循环纤维细胞到肺损伤部位,刺激上皮基质转化和成纤维细胞分化为肌成纤维细胞,促进成纤维细胞和肌成纤维细胞灶的形成。肌成纤维细胞增生分泌过量的细胞外基质,导致纤维瘢痕及蜂窝肺形成、肺结构破坏和功能丧失。
流行病学 IPF确切的患病率和发病率尚不清楚。IPF的患病率及发病率远比以前估计得高,同时死亡率也呈上升趋势。Raghu等调查了美国1996-2000年IPF的发病情况,估计患病率和发病率分别波动于(14.0~42.7)/10万人和(6.8~16.3)/10万人,认为IPF患病率较前很可能增加。欧洲几个国家的研究报告,IPF患病率(1~4)/10万人。在美国1992-2003年的IPF的死亡率为50.8/100万人,与前相比,男性增加28.4%,女性增加41.3%。在英格兰和威尔士,由IPF导致的死亡较过去20年已有3倍的增加。该病老年患者常见,诊断时平均年龄67岁,60%的患者年龄超过60岁。男性与女性患病率比例1.4:1.0,发病率男性与女性比例1.3:1.0。既往有吸烟史患者略多。
病理表现 大体病理:肺容积缩小,质地偏韧硬,脏层胸膜可见局限性瘢痕。切面观,弥漫性实变区和相对正常的肺结构相间存在,依疾病轻重不同其比例各异,严重受累处可见蜂窝肺。
组织病理学表现为UIP,成纤维细胞灶是其重要的特征性所见。
典型 UIP——满足以下4个条件:①明显的结构破坏和纤维化伴或不伴胸膜下蜂窝肺;②肺实质可见斑片状纤维化;③成纤维细胞灶;④无不支持UIP诊断的特征。
可能 UIP——满足以下3个条件:①明显的结构破坏和纤维化伴或不伴胸膜下蜂窝肺;②仅有斑片状纤维化和成纤维细胞灶所见之一者;③无不支持 UIP 诊断的特征。
疑似 UIP——满足以下3个条件:①斑片或弱漫的肺实质纤维化,伴或不伴肺间质炎症;②缺乏 UIP其他诊断条件;③无不支持UIP诊断的特征。
不支持UIP诊断的组织病理学所见主要有:透明膜形成、机化性肺炎、肉芽肿、远离蜂窝区明显的炎性细胞浸润、气道中心型病变等。
临床表现 多于50岁以后发病,呈隐匿起病,主要表现为活动性呼吸困难,渐进性加重,常伴干咳。全身症状不明显,可以有不适、乏力和体重减轻等,但很少发热。75%有吸烟史。
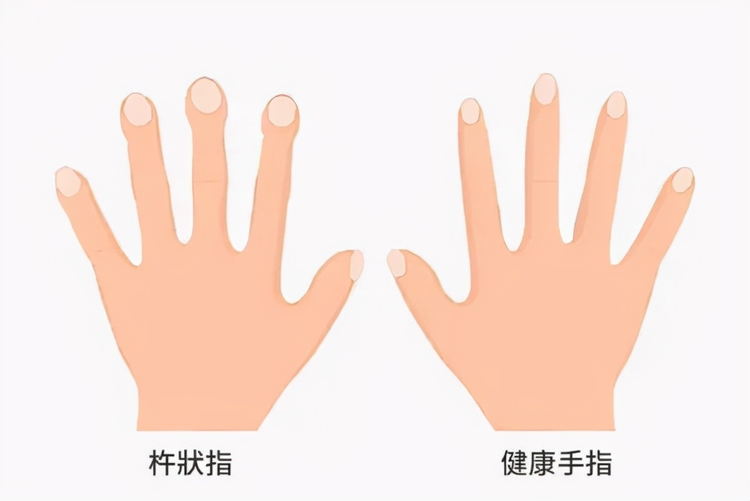
约半数病人可见杵状指(如下图),90%的病人可在双肺基底部闻及吸气末细小的Velcro啰音。在疾病晚期可出现明显发绀、肺动脉高压和右心功能不全征象。
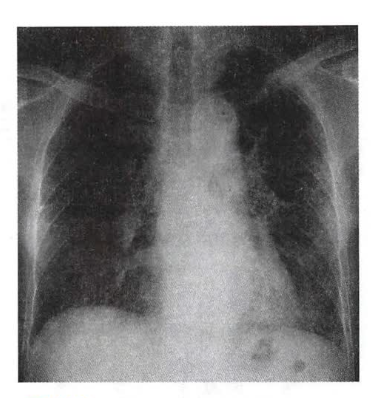
检查诊断 检查项目 X线检查:期常规胸部X线平片不能显示异常,随着病情的进展,95%的患者出现症状时均有胸片的异常,X线表现为云雾状、隐约可见微小点状的弥漫性阴影,犹如磨玻璃(如下图);进一步进展则纤维化表现愈加明显,从纤细的网织状到粗大的网织状,或成网织结节状,以双下肺和外周(胸膜下)明显;晚期更有大小不等的囊状改变,即蜂窝肺,肺容积缩小,膈肌上抬,叶间裂移位。
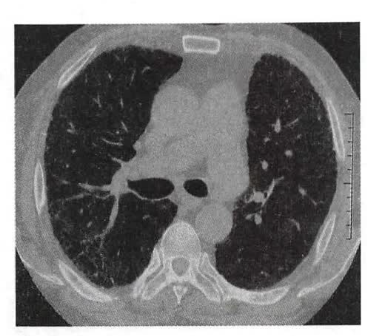
胸部高分辨率CT(HRCT):可以显示UIP的特征性改变(如下图),诊断UIP的准确性大于90%,因此HRCT已成为诊断IPF的重要方法,可以替代外科肺活检。HRCT的典型UIP表现为:①病变呈网格改变,蜂窝改变伴或不伴牵拉支气管扩张;②病变以胸膜下、基底部分布为主。
肺功能:主要表现为限制性通气功能障碍、弥散量降低伴低氧血症或Ⅰ型呼吸衰竭。早期静息肺功能可以正常或接近正常,但运动肺功能表现P(A-a) O2 增加和氧分压降低。
血液化验:血液涎液化糖链抗原(KL-6)增高,ESR、抗核抗体和类风湿因子可以轻度增高,但没有特异性。结缔组织疾病相关自身抗体检查有助于IPF的鉴别。
支气管肺泡灌洗液(BALF)/肺活检术(TBLB):BALF细胞分析多表现为中性粒细胞和(或)嗜酸性粒细胞增加。BAL或TBLB对于IPF无诊断意义。
外科肺活检:对于HRCT呈不典型UIP改变,诊断不清楚,没有手术禁忌证的病人应该考虑外科肺活检。IPF的组织病理类型是UIP,UIP的病理诊断标准为:①明显纤维化/结构变形伴或不伴蜂窝肺,胸膜下、间质分布;②斑片肺实质纤维化;③成纤维细胞灶。
诊断标准 IPF诊断遵循如下标准:①间质性肺疾病(ILD),但排除了其他原因(如环境、药物和结缔组织疾病等);②HRCT表现为UIP型;或③联合HRCT和外科肺活检病理表现诊断UIP。
IPF急性加重(acute exacerbation of IPF,AE-IPF):IPF病人出现新的弥漫性肺泡损伤导致急性或显著的呼吸困难即为AE-IPF。诊断标准:①过去或现在诊断IPF;②1个月内发生显著的呼吸困难加重;③CT表现为UIP背景下出现新的双侧磨玻璃影伴或不伴实变影;④不能完全由心衰或液体过载解释。
鉴别诊断 IPF的诊断需要排除其他病因的ILD。UIP是诊断IPF的金标准,但UIP也可见于慢性过敏性肺炎、石棉沉着病、结缔组织病(CTD)等。
过敏性肺炎多有环境抗原暴露史(如饲养鸽子、鹦鹉等),BAL细胞分析显示淋巴细胞比例增加。
石棉沉着病、硅沉着病或其他职业尘肺多有石棉、二氧化硅或其他粉尘接触史。
CTD多有皮疹、关节炎、全身多系统累及和自身抗体阳性。
治疗 IPF的治疗仍然是临床医师面临的难题。除肺移植能延长严重IPF生存期及改善患者的生活质量外,现有的药物能延缓肺功能下降速度,但不能阻止病情进展。需通过氧疗,肺康复等支持和对症处理改善患者生活质量;关注IPF急性加重、胃食管返流、肺动脉高压等常见并发症评价和处理。
药物治疗 自2011年《特发性肺纤维化诊断和治疗指南》发布以来,有关IPF药物治疗相关的临床研究有了重要进展。2015年美国胸科学会(ATS)/欧洲呼吸学会(ERS)/日本呼吸学会(JRS)/拉丁美洲胸科学会(ALAT)对2011年《特发性肺纤维化诊断和治疗指南》的治疗进行了更新(见下表),对轻度至中度肺功能下降IPF患者,推荐酌情给予吡非尼酮和尼达尼布药物治疗。
2011年与2015年《特发性肺纤维化诊断和治疗指南》药物治疗对比药物
2011年指南
2015年指南
糖皮质激素
强不推荐
强不推荐
糖皮质激素+硫唑嘌呤
强不推荐
强不推荐
抗凝药物(华法林)
弱不推荐
强不推荐
激素+N-乙酰半胱氨酸+硫唑嘌呤
弱不推荐
强不推荐
选择性ETA受体拮抗剂安贝坦生
未提及
强不推荐
单靶点酪氨酸激酶抑制剂伊马替尼
未提及
强不推荐
多靶点酪氨酸激酶抑制剂-尼达尼布
未提及
弱推荐(酌情推荐)
吡非尼酮
弱不推荐
弱推荐(酌情推荐)
波生坦
强不推荐
弱不推荐
西地那非
未提及
弱不推荐
抑酸治疗
弱推荐
弱推荐
N-乙酰半胱氨酸
弱不推荐
弱不推荐
对IPF-PH进行抗PH治疗
弱不推荐
弱不推荐
吡非尼酮:吡非尼酮(pirfenidone,PD)为化学合成物,化学名称5-甲基-1-苯基-H-吡啶-2酮。它是一种具有抗纤维化、抗氧化、抗炎等作用的小分子化合物。吡非尼酮按剂量递增原则逐渐增加用量,该药的初始用量为每次200mg,每日三次,温水送服剂;希望能在2周的时间内,通过每次增加200mg剂量,最后将该药用量维持在每次600mg(每日1800mg);使用时应密切观察患者用药耐受情况,若出现明显胃肠道症状、对日光或紫外线灯的皮肤反应、肝功能酶学指标的显著改变和体重减轻等现象时,可根据临床症状减少用量或者停止用药。在症状减轻后,可再逐步增加给药量,最好将维持用量调整在每次400mg(每日1200mg)以上。服用吡非尼酮期间,应定期(3-6个月)肺功能检查。《NICE(英国国立健康与临味优化研究所)指南》建议,如果患者肺功能在1年的治疗时间内,较基线时降低了10%或更多,应停止其吡非尼酮治疗。
尼达尼布:尼达尼布(nintedanib)是小分子三体酪氨酸激酶抑制剂,具有阻断血小板源性生长因子受体(PDGFR),成纤维细胞生长因子受体(FGFR),血管内皮细胞生长因子受体(VEGFR)的作用。在2期临床试验中发现,尼达尼布150mg,2次/天,有延缓患者FVC的下降趋势。
急性加重药物治疗:虽然强不推荐单用激素治疗 IPF患者,但对于急性加重IPF患者《2015年治疗指南》更新中,弱推荐给予大剂量激素治疗。可静脉甲强龙500-1000mg/d,3天后,改1mg/(kg•d)泼尼松或等效剂量激素继续治疗4-8周,根据患者病情和效果逐步减至维持量。急性加重初始治疗可先应给予广谱抗生素,直至感染被排除。环磷酰胶、硫唑嘌呤、环孢素A等免疫抑制剂治疗IPF急性加重效果不肯定。
其他药物:以往临床曾经使用的糖皮质激素、秋水仙碱、环孢素A、激素联合免疫抑制剂、干扰素(IFN)-
1b、泼尼松联合硫唑嘌呤和乙酰半胱氨酸,及抗凝药物(华法林)、波生坦和依那西普等药物列入强不推荐。非药物治疗 虽然吡非尼酮与尼达尼布已经用于IPF临床治疗,其效果难以令人满意,药物费用的昂贵也限制了其临床应用,因此,IPF治疗仍处于困境。针对每一具体IPF患者应积极地选择合适的支持及对症治疗,缓解患者临床症状,改善患者生活质量。
1.氧疗,在静息,睡眠,活动时维持患者脉搏血氧饱和度至少90%以上。吸氧可减轻运动所致的低氧血症,提高运动能力。
2.咳嗽是令部分IPF患者倍感痛苦的临床症状之一,口服可待因和其他镇咳药对有些患者可能有用。
3.肺康复治疗,包括患者评估、运动训练、教育、营养干预和社会心理支持等。稳定患者的心理;根据不同个体的情况制订合适的锻炼计划,有计划地安排日常活动,以维持患者的最佳骨骼肌肉状态,对将来的肺移植有益。
4.定期接种疫苗,预防肺炎和流感。
5.获得和维持理想的体重。
6.对所有的IPF患者进行肺移植评价,筛选合格的肺移植候选人,安排的合格候选人肺移植登记和等待。
7.对患者进行系列的肺生理,气体交换,运动能力和HRCT监测,为IPF的预后研究提供准确资料,以优化IPF今后临床处理的决策。
8.关注对胃食管反流,睡眠呼吸障碍,肺动脉高压,冠心病等常见合并症的进行评价和处理。
肺移植 肺移植是治疗IPF最有效的治疗方法。当肺功能严重不全,低氧血症持续迅速恶化,但无严重并发症,且年龄小于60岁,如有条件可考虑肺移植。肺移植可改善IPF患者的生活质量,5年生存率可达50%~56%。
预防 防治刺激性物质的长期吸入,如具有腐蚀性的化学性气体、煤尘、粉尘等。积极宣传呼吸道防护知识。
加强病人教育与自我管理,建议吸烟者戒烟,预防流感和肺炎。
定期接种疫苗,预防肺炎和流感。
预后 IPF诊断后中位生存期为2-3年,但IPF自然病程及结局个体差异较大。大多数病人表现为缓慢逐步可预见的肺功能下降;少数病人在病程中反复出现急性加重;极少数病人呈快速进行性发展。影响IPF 病人预后的因素包括:呼吸困难、肺功能下降和HRCT纤维化及蜂窝样改变的程度,6分钟步行试验(6MWT)的结果,尤其是这些参数的动态变化。基线状态下DLCO<40%预计值和6MWT时SpO2 <88%,6-12个月内FVC绝对值降低10%以上或DLCO绝对值降低15%以上都是预测死亡风险的可靠指标。
历史 随着对特发性间质性肺炎的认识深入,特发性肺纤维化(IPF)的概念及内在含义历经变迁。在Liebow的博性间质性肺炎病理分类中,首次提出了UIP的概念,pneumonia 描述非感染性炎症,“usual”意指最常观察到的组织病理表现的类型,UIP仅仅是一个病理学的诊断术语,而非独立的疾病实体。IPF是一个临床分类的术语,以往将隐匿性致纤维化肺泡炎(cryptogenic fibrosing alveolitis)视为同义语。2000年前,以往文献提及的IPF,曾包含数种不同病理类型的间质性肺炎,其临床病程和预后各不相同。2000年美国胸科学会(ATS)和欧洲呼吸学会(ERS)发表了有关IPF诊断和治疗的多国专家共识中,对 IPF 做了重新的界定。IPF 被定义为一种原因不明,组织病理学表现为普通型间质性肺炎(UIP),局限于肺部的慢性致纤维化型间质性肺炎。2011年3月ATS、ERS、JRS和ALAT颁布《特发性肺纤维化诊断和治疗指南》,将IPF的定义为原因不明,成人(多为老年),局限于肺,进行性致纤维化的间质性肺炎,其组织病理学和或放射学表现为UIP型。《IPF指南》首次将放射学 UIP 型表现写入IPF 的定义中,强调识别高分辨率 CT(HRCT)UIP 型的重要性及诊断作用。