进行性脊髓性肌萎缩症是一类累及脊髓前角运动神经元和脑干运动神经核的疾病。病因尚未明确。大多数学者认为是常染色体隐性遗传,小部分为基因突变引起。临床表现为进行性、对称性,肢体近端为主的广泛性弛缓性麻痹与肌萎缩。智力发育及感觉均正常。本病尚无特异的有效治疗,主要治疗措施为对症治疗。预后主要与疾病的类型有关,总体来说,预后较差。
本页面主要目录有关于进行性脊肌萎缩症的:就诊科室、病因、症状、检查、诊断、鉴别诊断、治疗、危害、预后、预防等介绍

新亚洲娱乐(英文名:ASIA ENTERTAINMENT GROUP LIMITED,全称:新亚洲娱乐联盟集团有限公司)是一家以从事戏剧制作人及杂项戏剧服务为主的企业,成立于1999年,位于香港特别行政区。旗下分公司包括虎威艺能创作有限公司(TGS HK)、稻草人娱乐创作社(Scarecrow Entertainment)、虎威王朝音乐创作股份有限公司(TGS Music)、虎威活力娱乐传播有限公司(TGS Taiwan)、AK Entertainment(Korea)以及AEG Korea等。
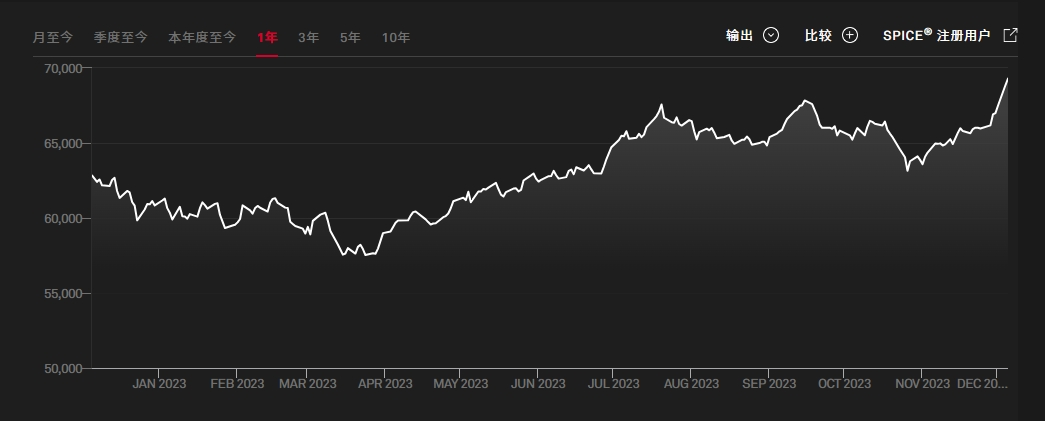
印度孟买SENSEX30指数(又称孟买敏感指数)为印度最被广泛使用的指数,为投资印度的重要参考指标,是由孟买证券交易所发行。由于各类媒体提到的“印度股市”,实际上都是孟买股票交易所,因此,该交易所的SENSEX-30指数几乎成了印度股市的代名词。




