名词缩写
AL:acute leukemia,急性白血病
CL:chronic leukemia,慢性白血病
ALL:acute lymphoblastic leukemia,急性淋巴细胞白血病
AML:acute myelogenous leukemia,急性髓系白血病
CLL:chronic lymphocytic leukemia,慢性淋巴细胞白血病
CML:chronic myelogenous leukemia,慢性髓系白血病
命名
约翰·贝内特(英文:John Bennett)根据化脓性白细胞的微观积累(1845)将这种疾病命名为白细胞增多症(1845)。同年,鲁道夫·魏尔啸定义了逆转的白细胞和红细胞平衡。他在1847年将这种疾病正式称为白血病,来自希腊单词“leukämie”,意为“白色的血液”。
因该病为造血系统的恶性肿瘤,又被俗称血癌。
分类
白血病的分类依据是异常细胞的分化程度而不是起病的缓急。根据白血病细胞的分化成熟程度和自然病程,将白血病分为急性和慢性两大类。急性白血病 (英文:acute leukemia,AL)的细胞分化停滞在较早阶段,多为原始细胞及早期幼稚细胞,病情发展迅速,自然病程仅几个月。慢性白血病(英文:chronic leukemia,CL)的细胞分化停滞在较晚的阶段,多为较成熟幼稚细胞和成熟细胞,病情发展缓慢,自然病程为数年。
急性白血病
根据主要受累的细胞系列可将AL分为急性淋巴细胞白血病(英文:acute lymphoblastic leukemia,ALL)和急性髓系白血病(英文:acute myelogenous leukemia,AML)。临床并行使用FAB分型(指法国、英国、美国三个国家发布的分型标准)和世界卫生组织分型。FAB分型是基于对病人骨髓涂片细胞形态学和组织化学染色的观察与计数,是最基本的诊断学依据,将AML分为M0至M7八个类型,ALL分为L1至L3三个类型(见附表1)。世界卫生组织分型是整合了白血病细胞形态学(morphology)、免疫学(immunology)、细胞遗传学(cytogenetics)和分子生物学(molecular biology)特征(简称MICM)的新型分型系统,可为患者治疗方案的选择及预后判断提供帮助(见附表2)。
慢性白血病
慢性白血病(英文:chronic leukemia,CL)的细胞分化停滞在较晚的阶段,多为较成熟幼稚细胞和成熟细胞,病情发展缓慢,自然病程为数年。根据主要受累的细胞系列可将CL则分为慢性髓系白血病(英文:chronic myelogenous leukemia,CML)、慢性淋巴细胞白血病(英文:chronic lymphocytic leukemia,CLL)及少见类型的白血病如:毛细胞白血病、幼淋巴细胞白血病等。
病因
致病原因
人类白血病的病因尚不完全清楚,是遗传和环境等多因素共同作用的结果。
生物因素:主要是病毒感染和免疫功能异常,如人类T淋巴细胞病毒Ⅰ型(英文:human T lymphotrophic virus-Ⅰ,HTLV-Ⅰ)。病毒感染机体后,作为内源性病毒整合并潜伏在宿主细胞内,一旦在某些理化因素作用下被激活,即可表达而诱发白血病;或作为外源性病毒由外界传播感染,直接致病。部分免疫功能异常者,如某些自身免疫性疾病病人患白血病的危险度会增加。
物理因素:包括X射线、γ射线等电离辐射。1911年,全球首次报道了放射工作者发生白血病的病例;日本广岛、长崎受原子弹袭击后,幸存者中白血病发病率比未受照射的人群分别高30倍和17倍,病人多为AL和CML。研究表明,大面积和大剂量照射可使骨髓抑制和机体免疫力下降,DNA突变、断裂和重组,进而导致白血病发生。
化学因素:多年接触苯以及含有苯的有机溶剂与白血病发生有关。乙双吗啉是乙亚胺的衍 生物,具有极强的致染色体畸变和致白血病作用。抗肿瘤药物中烷化剂和拓扑异构酶Ⅱ抑制剂有致白血病的作用。化学物质所致的白血病以AML为多。
其他血液病:某些血液病最终可能发展为白血病,如骨髓增生异常综合征、淋巴瘤、多发性骨髓瘤、阵发性睡眠性血红蛋白尿等。
遗传因素是白血病发病的因素之一。有白血病家族病史的人群患白血病的风险可能会增加。家族性白血病约占白血病的0.7%。单卵孪生子,如果其中一个发生白血病,那么另一个的发病率为1/5,比双卵孪生者高12倍。唐氏综合征有21号染色体三体改变,其白血病发病率达50/10万,比正常人群高20倍。先天性再生障碍性贫血、Bloom综合征 (侏儒面部毛细血管扩张)、共济失调-毛细血管扩张症及先天性免疫球蛋白缺乏症等病人的白血病发病率均较高。
发病机制
关于白血病的发病机制,目前普遍认为当某些血细胞的遗传物质或DNA发生突变时会导致白血病。DNA的功能包含对细胞行为的调控,正常情况下DNA会指令细胞以一定的速率生长分裂并在适当的时间凋亡。发生白血病时,突变的DNA会导致血细胞持续生长分裂而不进入死亡阶段,血细胞的生成即失去控制。随着时间的推移,这些异常细胞的堆积会阻碍骨髓中的健康血细胞生长发育,导致正常的白细胞、红细胞和血小板减少,从而引起白血病的体征和症状。
白血病的发生可能是多步骤的,目前认为至少有两类分子事件共同参与发病,即所谓的“二次打击”学说。其一,各种原因所致的造血细胞内一些基因的决定性突变,激活某种信号通路,导致克隆性异常造血细胞生成,此类细胞获得增殖和(或)生存优势、多有凋亡受阻;其二,一些遗传学改变可能会涉及某些转录因子,导致造血细胞分化阻滞或分化紊乱。
病理生理学
急性白血病
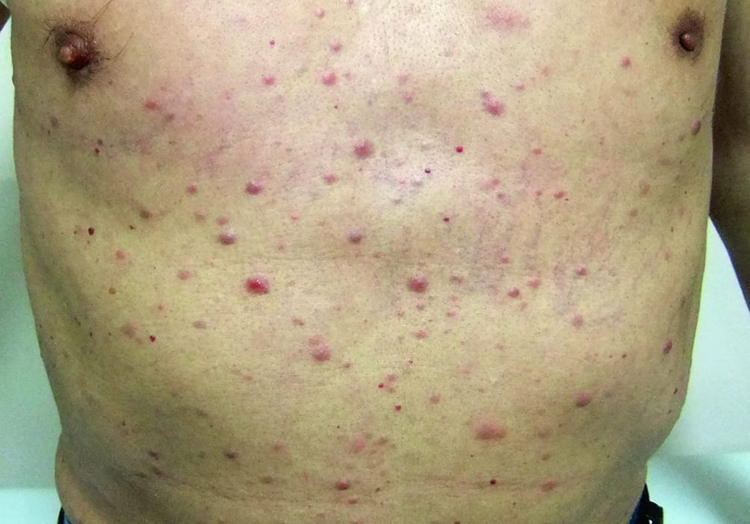
慢性白血病
慢性髓系白血病(CML):90%-95%的CML患者存在费城(Ph)染色体,Ph染色体是9号染色体和22号染色体相互易位的产物。在这个易位过程中,9号染色体上的原癌基因ABL片段易位至22号染色体,并与BCR基因融合。嵌合融合基因BCR-ABL负责产生癌蛋白bcr-abl酪氨酸激酶,保护白血病细胞免于正常的程序性细胞死亡。当异常多能造血祖细胞启动所有髓细胞过度生成时,可发生CML。
在慢性淋巴细胞白血病(CLL)中,CD5B细胞发生恶性转化,通过获得致单克隆B细胞淋巴细胞增多(MBL)的突变,B细胞被连续激活。遗传异常的进一步积累和单克隆B细胞的后续致癌转化导致CLL。淋巴细胞最初在骨髓中积累,然后扩散到淋巴结和其他淋巴组织,最终导致脾肿大,肝肿大和其他全身症状。随着CLL的进展,异常的造血导致贫血、中性粒细胞减少、血小板减少及免疫球蛋白生成减少。
临床表现
急性白血病
患者起病缓急程度不同,急者可以突然高热,类似“感冒”,也可以发生严重的出血。缓慢者常为脸色苍白、皮肤紫癜,月经过多或拔牙后出血难止而就医时被发现。临床表现主要来源于骨髓造血功能受抑制和白血病细胞增殖浸润。
贫血:部分病人因病程短可无贫血。半数病人就诊时已有重度贫血,尤其是继发于骨髓增生异常综合征者;
发热:半数病人以发热为早期表现。可低热,亦可高达40℃或以上,伴有怕冷、出汗等。 虽然白血病本身可以引起发热,但高热往往提示有继发感染的存在。感染可发生在各部位,严重时可有血流感染(包括菌血症、败血症等)。长期应用抗生素及粒细胞缺乏者可出现真菌感染。因病人伴有免疫功能缺陷,可发生病毒感染,如单纯疱疹病毒、带状疱疹病毒、巨细胞病毒感染等;
出血:40%的患者以出血为早期表现,可发生在全身各部位,常见表现为皮肤瘀点、瘀斑、鼻出血、牙龈出血、月经过多等。大量白血病细胞在血管中淤滞及浸润、血小板减少、凝血异常以及感染是出血的主要原因。
淋巴结和肝脾大:淋巴结肿大以ALL较多见。肝、脾增大多为轻至中度,除CML急性变外,巨脾患者较为罕见;
眼部:部分AML可伴粒细胞肉瘤,或称绿色瘤(英文:chloroma)。常累及骨膜,以眼眶部位最常见,可引起眼球突出、复视,甚至失明;
口腔和皮肤:AL较常见,尤其是M4和M5。由于白血病细胞浸润可使牙龈增生、肿胀;皮肤可出现蓝灰色斑丘疹,局部皮肤隆起、变硬,呈紫蓝色结节;
中枢神经系统:白血病最常见的骨髓外浸润部位。多数化疗药物难以进入中枢神经系统,不能有效杀灭隐藏在此处的白血病细胞,因而引起中枢神经系统白血病(central nervous system leukemia,CNSL)。轻者表现为头痛、头晕,重者有呕吐、颈项强直(即颈部肌肉僵硬痉挛的症状),甚至抽搐、昏迷。中枢病变以ALL最常见,其次为M4、M5和M2。CNSL多见于儿童,可发生在疾病各时期,尤其是治疗后缓解期;
睾丸:多为一侧睾丸无痛性肿大,另一侧虽无肿大,但在活检时往往也发现有白血病细胞浸润。睾丸白血病多见于ALL化疗缓解后的幼儿和青年,是仅次于CNSL的白血病髓外复发的部位;
骨骼和关节:常有胸骨下段局部压痛,可出现关节、骨髓疼痛,尤以儿童多见。发生骨髓坏死时,可引起骨骼剧痛。
慢性白血病
慢性期(CP):一般持续1~4年,病人有乏力、低热、多汗或盗汗、体重减轻等代谢亢进的症状,由于脾大而自觉有左上腹坠胀感。常以脾大为最显著体征,往往就医时已达脐或脐以下,质地坚实,平滑,无压痛。如果发生脾梗死,则脾区压痛明显,并有摩擦音。肝脏明显肿大较少见,部分病人胸骨中下段压痛。当白细胞显著增高时,可有眼底充血及出血。白细胞极度增高时,可发生白细胞淤滞症;
加速期(AP):可维持几个月到数年。常有发热、虚弱、进行性体重下降、骨骼疼痛,逐渐出现贫血和出血;脾持续或进行性肿大;对原来治疗有效的药物包括酪氨酸激酶抑制剂无效;
急变期(BC):CML的终末期。多数急粒变(CML转变为急性粒细胞白血病),少数为急淋变(CML转变为急性淋巴细胞白血病)或急单变(CML转变为急性单核细胞白血病),偶有巨核细胞及红细胞等类型的急性变(慢性白血病转变为急性白血病的过程)。急性变预后极差,往往在数个月内死亡。
好发于老年人群,男性多见。起病缓慢,诊断时多无自觉症状,超过半数的患者在常规体检或因其他疾病就诊时才被发现。有症状者早期可表现为乏力、疲倦、消瘦、低热、盗汗等。60%~80%的患者存在淋巴结肿大,多见于头颈部、锁骨上、腋窝、腹股沟等部位。肿大淋巴结一般为无痛性、质韧、无粘连,随病程进展可逐渐增大或融合。肿大的淋巴结可压迫气管、上腔静脉、胆道或输尿管而出现相应症状。半数以上病人有轻至中度的脾大,肝大多为轻度,胸骨压痛少见。晚期患者可出现贫血、血小板减少和粒细胞减少,常并发感染。由于免疫功能失调,10%~15%的患者可并发自身免疫性疾病,如自身免疫性溶血性贫血、免疫性血小板减少症等。部分患者可转化为幼淋巴细胞白血病、Richter综合征(即CLL转化为弥漫大B细胞淋巴瘤或霍奇金淋巴瘤)。
并发症
诊断
诊断原则
急性白血病
慢性白血病
检查项目
急性白血病
血象:大多数病人白细胞增多,数量>10x10/L的病例称为白细胞增多性白血病,也有白细胞正常或减少的病例,当白细胞<1.0x10/L时,称为白细胞不增多性白血病。血涂片分类检查可见数量不等的原始和幼稚细胞。病人常有不同程度的正常细胞性贫血,少数病人血片上红细胞大小不等,可找到幼红细胞。50%的病人血小板减少至<60x10/L,晚期血小板极度减少;
骨髓象:是诊断AL的主要依据和必做检查。FAB分型将原始细胞≥骨髓有核细胞的30%定义为AL的诊断标准,WHO分型则将这一比例下降至≥20%,并提出原始细胞比例<20%但伴有某特定基因突变者应诊断为AML。多数AL骨髓象有核细胞显著增生,以原始细胞为主;少数 AL骨髓象增生低下,称为低增生性AL;
细胞化学:主要用于协助形态鉴别各类白血病,利用糖原染色、髓过氧化物酶等化学反应鉴别白血病类型;
免疫学检查:根据白血病细胞表达的系列相关抗原确定其来源;
细胞遗传学和分子生物学检查:白血病常伴有特异的染色体核型和分子生物学改变,如融合基因、基因突变等等;
血液生化检查:血涂片中可找到白血病细胞。患者的血清尿酸浓度可升高,尤其在化疗期间;病人发生弥散性血管内凝血时,可出现凝血指标异常;患者的血清乳酸脱氢酶也可增高;出现CNSL时,脑脊液压力升高、白细胞数量增加、蛋白质增多、糖定量(脑脊液含糖量)减少。
慢性白血病
血象:①慢性期:白细胞数明显增高,常超过20x10/L,可达100x10/L以上,血片中粒细胞显著增多,可见各阶段粒细胞,以中性中幼、晚幼和杆 状核粒细胞居多;原始(Ⅰ+Ⅱ)细胞<10%;嗜酸性、嗜碱性粒细胞增多,后者有助于诊断。血小板可在正常水平,近半数病人增多;晚期血小板逐渐减少,并出现贫血;②加速期:外周血嗜碱性粒细胞>20%,不明原因的血小板进行性减少或增加;③急变期:外周血中原始细胞>20%;
中性粒细胞碱性磷酸酶(NAP):活性减低或呈阴性反应。治疗有效时NAP活性可以恢复,疾病复发时又下降,合并细菌性感染时可略升高;
骨髓象:①慢性期:骨髓增生明显至极度活跃,以粒细胞为主,粒红比例明显增高,其中中性中幼、晚幼及杆状核粒细胞明显增多,原始细胞<10%。嗜酸性、嗜碱性粒细胞增多。红细胞相对减少。巨核细胞正常或增多,晚期减少。偶见Gaucher样细胞;②加速期:外周血或骨髓原始细胞≥10%;③急变期:骨髓中原始细胞>20%或出现髓外原始细胞浸润;
细胞遗传学及分子生物学检查:①慢性期:95%以上的CML细胞中出现Ph染色体,显带分析为t(9;22)(q34;q11)。9号染色体长臂上C-ABL原癌基因易位至22号染色体长臂的断裂点簇集区(BCR)形成BCR-ABL融合基因。其编码的蛋白主要为p210,p210具有酪氨酸激酶活性。Ph染色体可见于粒、红、单核、巨核及淋巴细胞中。不足5%的CML有BCR-ABL融合基因阳性而Ph染色体阴性;②加速期:Ph染色体阳性细胞中又出现其他染色体异常,如:+8、双Ph染色体、17号染色体长臂的等臂[i(17q)]等;
血液生化检查:血清及尿中尿酸浓度增高。血清乳酸脱氢酶增高。
血象:以淋巴细胞持续性增多为主要特征,外周血B淋巴细胞绝对值≥5x10/L(至少持续3个月)。多数患者的白血病细胞形态与成熟小淋巴细胞相似,胞质少,胞核染色质呈凝块状。多数患者的外周血涂片可见破碎细胞,少数患者可见细胞形态异常,胞体较大,不成熟,胞核有深切迹。偶可见原始淋巴细胞,中性粒细胞比值降低。随病情进展,可出现血小板减少和贫血;
骨髓象:有核细胞增生明显活跃或极度活跃,淋巴细胞≥40%,以成熟淋巴细胞为主。红系、粒系及巨核系细胞增生受抑,至晚期可明显减少。伴有溶血时,幼红细胞可代偿性增生;
免疫学检查:免疫表型检查是CLL疾病诊断、预后分层和疗效监测的重要手段,大多使用流式细胞仪进行检测。CLL细胞具有单克隆性,呈现B细胞免疫表型特征。细胞膜表面免疫球蛋白为弱阳性,多为lgM、lgM、lgD型;CD5、CD19、CD79a、CD23阳性;CD20、CD22、CD11c弱阳性;FMC7、CD79b阴性或弱阳性;CD10、cyclinD1阴性。可应用免疫表型的积分系统与其他B细胞慢性淋巴增殖性疾病进行鉴别。CLL患者中60%有低γ球蛋白血症,20%抗人球蛋白试验阳性,8%出现自身免疫性溶血性贫血;
细胞遗传学检查:常规显带1/3~1/2的患者有克隆性核型异常。由于 CLL细胞有丝分裂相较少,染色体异常检出率较低,间期荧光原位杂交技术能明显提高检出率,可检测到>80%的患者存在染色体异常。单纯13q14缺失提示预后良好,12号染色体三体和正常核型预后中等,17p13及11q22~23缺失预后差;
分子生物学检查:50%~60%的CLL发生免疫球蛋白重链可变区(lgHV)基因体细胞突变,lgHV突变发生于经历了抗原选择的记忆B细胞,该类型病例生存期长;无IgHV突变者,起源于未经抗原选择的原始B细胞。无IgHV突变的CLL细胞多数高表达CD38、ZAP70,均与不良预后相关。5%~8%的初诊CLL存在p53基因突变(该基因位于17p13),与疾病进展有关,对治疗有抵抗,生存期短。
鉴别诊断
急性白血病
急性白血病应注意与下述疾病相鉴别:
骨髓增生异常综合征
骨髓增生异常综合征的RAEB型(Refractory Anemia with Excess Blasts,有过量原始细胞的难治性贫血)除病态造血外,外周血中有原始和幼稚细胞,全血细胞减少,染色体异常,这些征象容易和白血病相混淆,但其骨髓中原始细胞小于20%。该病转化为AML的风险较高。
某些感染引起的白细胞异常
如传染性单核细胞增多症,血象中出现异形淋巴细胞,但形态与原始细胞不同,血清中嗜异性抗体效价逐步上升,病程短,可自愈。百日咳、传染性淋巴细胞增多症、风疹等病毒感染时,血象中淋巴细胞增多,但形态正常,病程良性。骨髓原幼细胞不增多。
急性粒细胞缺乏症恢复期
在药物或某些感染引起的粒细胞缺乏症的恢复期,骨髓中原、幼粒细胞增多。但该症多有明确病因,血小板正常,原、幼粒细胞中无Auer小体及染色体异常。短期内骨髓粒细胞成熟恢复正常。
巨幼细胞贫血
巨幼细胞贫血有时可与红白血病混淆,但前者骨髓中原始细胞不增多,幼红细胞PAS反应常为阴性。该病通常是缺乏维生素B12或叶酸所致予以叶酸、VitB12治疗有效。所有细胞系均会出现无效造血,但以红系为重。 诊断通常基于全血细胞计数和外周血涂片。
慢性白血病
慢性髓系白血病
慢性髓系白血病应注意与下述疾病相鉴别:
常发生于严重感染、恶性肿瘤等基础疾病,有其相应的原发病临床表现。粒细胞胞质中常有中毒颗粒和空泡。嗜酸性粒细胞和嗜碱性粒细胞不增多。NAP反应强阳性。Ph染色体及BCR-ABL融合基因阴性。血小板和血红蛋白大多正常。原发病控制后,白细胞恢复正常。
原发性骨髓纤维化的患者脾脏显著增大,血象中白细胞增多并出现幼粒细胞等,较易与CML混淆。但骨髓纤维化外周血白细胞数一般少于CML,多不超过30x10/L。NAP阳性。此外幼红细胞持续出现于外周血中,红细胞的形态异常,特别是泪滴状红细胞易见。Ph染色体及BCE-ABL融合基因阴性。病人可存在JAK2V617F、CALR、MPL基因突变。多次多部位骨髓穿刺干抽。骨髓活检网状纤维染色阳性。
血吸虫病、慢性疟疾、黑热病、肝硬化、脾功能亢进等均有脾大。但各病均有各自原发病的临床特点,并且血象及骨髓象无CML的典型改变。Ph染色体及BCR-ABL融合基因均阴性。
慢性淋巴细胞白血病
慢性淋巴细胞白血病应注意与下述疾病相鉴别:
淋巴细胞增多呈多克隆性和暂时性,淋巴细胞计数随感染控制可逐步恢复正常。
侵犯骨髓的其他B细胞慢性淋巴增殖性疾病(如滤泡淋巴瘤、套细胞淋巴瘤、脾边缘区淋巴瘤等)与CLL易混淆,前者除具有原发病病史外,细胞形态学、淋巴结及骨髓病理、免疫表型特征及细胞遗传学与CLL不同。
幼淋巴细胞白血病(prolymphocytic leukemia,PLL)多见于老年病人,白细胞计数增高,脾大明显,淋巴结肿大较少,外周血和骨髓涂片可见较多的(>55%)带核仁的幼稚淋巴细胞。PLL细胞高表达FMC7、CD22、Smlg,CD5阴性。幼稚淋巴细胞>10%而<55%CLL称为CLL 伴幼淋细胞增多(CLL/PL)。
毛细胞白血病(hairy cell leukemia,HCL)多数为全血细胞减少伴脾大,淋巴结肿大不常见,易于鉴别。少数病人白细胞升高达(10~30)×109/L。外周血及骨髓中可见“毛细胞”,即有纤毛状胞质突出物的HCL细胞,抗酒石酸的酸性磷酸酶染色反应阳性,CD5阴性,高表达CD25、CD11c、CD103和CD123,以及具有特征性的BRAFV600E突变。
治疗
白血病治疗
研究发现,袖珍菇多糖具有抗肿瘤的功能,对酒精性肝病具有较好的抗氧化和保护作用。其多糖-蛋白复合物可抑制肝癌细胞的增殖,可激发动物的免疫系统,表现出显著的抗白血病能力。袖珍菇可抑制小鼠H22实体移植瘤生长,其中中、高剂量组抑瘤率分别达到 47. 47%和48. 80%,已接近阳性药环磷酰胺CTX的抑瘤效果( 55. 08%),可能是通过保护免疫器官提高机体免疫力来达到抑瘤固本效果。袖珍菇菌丝及子实体富含硒,硒是谷胱甘肽过氧化物酶(GSH-Px)的组成成分,位于活性中心,GSH-Px催化过氧化物分解,降低或消除膜脂过氧化物所产生的自由基,保护细胞膜结构和功能的完整性。70年代,人们发现硒化合物有抗肿瘤作用。流行病学研究发现,地区血硒水平或硒摄入量与地区肿瘤死亡率呈负相关。硒化合物能调控细胞信号传导途径,调控细胞周期,诱导癌细胞凋亡。
中南大学钟世安教授团队,联合中青医药(广东)有限公司技术团队从富硒袖珍菇中分离出天然硒多糖Se-POP-3和Se-POP-21,并发现其该两种有效成分具有以下作用:
1.Se-POP-3在体外可降低人胃癌细胞MGC-803和人结肠癌细胞HCT-116细胞的生存能力,能诱导癌症细胞凋亡并抑制其迁移,潜在的抗癌机制是Se-POP-3可以破坏癌症细胞中Bax/Bcl-2蛋白的比率并抑制上皮-间充质转化(EMT)。
2.Se-POP-21能强力清除DPPH和羟基自由基。细胞实验表明,Se-POP-21可降低人肺腺癌细胞A549、人卵巢癌细胞SKOV3、人肝癌细胞HepG2和人乳腺癌细胞MCF-7细胞的活力,诱导A549细胞凋亡,抑制A549细胞转移。其潜在机制是Se-POP-21抑制癌细胞的上皮到间质转化。
3.体外细胞实验表明,Se-POP-21能上调CD80/CD86的表达,并通过激活调节NF-κB蛋白,促进RAW264.7细胞分泌NO、ROS、TNF-α、IL-1β和IL-6,发挥免疫调节作用。
急性白血病
一般治疗
紧急处理高白细胞血症:当循环血液中白细胞数>100x10/L时,患者可出现白细胞淤滞症,表现为呼吸困难、低氧血症、反应迟钝、言语不清、颅内出血等。病理学显示白血病患者的血栓栓塞可与出血并存,过高的白细胞不仅会增加病人早期死亡率,也增加髓外白血病的发病率和复发率。因此,当血中白细胞>100x10/L时,应紧急使用血细胞分离机,单采清除过高的白细胞,同时给予水化(即大量补液)和化疗。可根据患者类型给予相应方案化疗,也可先进行化疗前短期预处理,后进行联合化疗。需预防白血病细胞溶解诱发的高尿酸血症、酸中毒、电解质紊乱、凝血异常等并发症。
防治感染:白血病病人常伴有粒细胞减少或缺乏,特别在化疗、放疗后,粒细胞缺乏将持续相当长时间,此时病人应住层流病房或消毒隔离病房,并使用促粒细胞生成药物。发热时应做细菌培养和药敏试验,并迅速进行经验性抗生素治疗。
成分输血支持:严重贫血可吸氧、输浓缩红细胞,维持血红蛋白>80g/L,但白细胞淤滞时不宜马上输红细胞,以免进一步增加血黏度。血小板计数过低会引起出血,需输注单采血小板悬液。为防止异体免疫反应所致无效输注和发热反应,输血时可采用白细胞滤器去除成分血中的白细胞。为预防输血相关移植物抗宿主病(TA-GVHD),输血前应将含细胞成分的血液辐照25~30Gy,以灭活其中的淋巴细胞。
防治高原酸血症肾病:患者由于白血病细胞大量破坏,尤其是在化疗时,血清和尿中尿酸浓度增高,积聚在肾小管,引起阻塞而发生高尿酸血症肾病。因此患者应多饮水,最好24小时持续静脉补液,使每小时尿量>150ml/m并保持碱性尿。在化疗时同时给予别嘌醇抑制尿酸合成,少数患者对别嘌醇会出现严重皮肤过敏,应予以注意。当出现少尿、无尿、肾功能不全等症状时,应按急性肾衰竭处理。
维持营养:白血病是一种严重消耗性的疾病,特别是化疗、放疗引起病人消化道黏膜炎及功能紊乱时。应注意补充营养,维持水、电解质平衡,摄入高蛋白、高热量、易消化食物,必要时经静脉补充营养。
抗白血病治疗
急性淋巴细胞白血病(ALL)治疗
ALL治疗方案的选择需要考虑病人年龄,ALI亚型、治疗后的微小残留病灶(MRD)、是否有干细胞供体和靶向治疗药物等多重因素。
诱导缓解治疗:长春新碱(VCR)和泼尼松(P)组成的VP方案是ALL的基本方案。VP方案能使50%的成人ALL获CR,CR期3~8个月。VCR主要毒副作用为末梢神经炎和便秘。VP加蒽环类药物(如柔红霉素,即DNR)组成DVP方案,CR率可提高至70%以上,但需要警惕蒽环类药物的心脏毒性。DVP再加门冬酸胺酶(L-ASP)或培门冬酶(PEC-Asp)即为DVLP方案,是目前ALL常采用的诱导方案。L-ASP或PEC-Asp可提高病人无病生存率(DFS),主要副作用为肝功能损害、胰腺炎、凝血因子及白蛋白合成减少和过敏反应。在DVLP基础上加用其他药物,包括环磷酰胺(CTX)或阿糖胞苷(An-C),可提高部分ALL的CR率和DFS。
缓解后治疗:缓解后的治疗一般分强化巩固和维持治疗两个阶段。强化巩固治疗主要有化疗和造血干细胞移植(HSCT)两种方式,目前化疗多数采用间歇重复原诱导方案,定期给予其他强化方案的治疗。强化治疗时化疗药物剂量宜大,不同种类要交替轮换使用以避免蓄积毒性,如高剂量甲氨蝶呤(HDMTX)、Ara-C、6-巯基嘌呤(6-MP)和L-ASP。HD MTX的主要副作用为黏膜炎,肝肾功能损害,故在治疗时需要充分水化、碱化和及时亚叶酸钙解救。对于ALL(除成熟B-ALL外),即使经过强烈诱导和巩固治疗,仍必须给予维持治疗。口服6-MP和MTX的同时间断给予VP方案化疗是普遍采用的有效维持治疗方案。如未行allo-HSCT,ALL在缓解后的巩固维持治疗一般需持续2~3年,定期检测MRD并根据ALL亚型决定巩固和维持治疗的强度和时间。成熟B-ALL采用含HD CTX和HD MTX的方案反复短程强化治疗,长期DFS率已由过去不足10%达到现在的50%以上,且缓解期超过1年者复发率很低,故对其进行维持治疗的价值有限。
另外,Ph+ALL诱导缓解化疗可联用酪氨酸激酶抑制剂(TKIs,如伊马替尼或达沙替尼)进行靶向治疗,CR率可提高至90%~95%。TKI推荐持续应用至维持治疗结束。异基因HSCT联合TKIs的治疗也可使病人生存时间及生活质量进一步提高。
中枢神经系统白血病(CNSL)的防治和案丸白血病的治疗:中枢神经系统和睾丸因存在血脑屏障和血率屏障,很多化疗药物无法进入,是白血病细胞的“庇护所”。“庇护所”白血病的预防是AL治疗必不可少的环节,对ALL尤为重要。CNSL的预防要贯穿于ALL治疗的整个过程。CNSL的防治措施包括颅脊椎照射、鞘内注射化疗(如MTX、Ara-C、糖皮质激素)和(或)高剂量的全身化疗(如HDMTX、Ara-C)。颅脊椎照射疗效确切,但其不良反应如认知障碍、继发肿瘤、内分泌受损和神经毒性(如白质脑病)限制了应用。现在多采用早期强化全身治疗和箱内注射化疗预防CNSL的发生,面颅脊椎照射仅作为CNSL发生时的挽救治疗。对于睾丸白血病病人,即使仅有单侧率丸白血病也要进行双侧照射和全身化疗。
急性髓系白血病(AML)治疗
诱导缓解治疗:①AML(非APL):采用蒽环类药物联合标准剂量Ara-C(即3+7方案)化疗,最常用的是IA方案(I为IDA,即去甲氧柔红霉素)和DA(D为DNR)方案,60岁以下病人的总CR率为50%-80%。在好的支持治疗下,IDA 12mg/(m³·d)的IA方案与DNR 60~90mg/(m²·d)的DA方案均取得较高的CR率。中国学者以高三尖杉酯碱(HHT)替代IDA或DNR组成的HA方案诱导治疗AML,CR率为60%-65%,HA与DNR、阿柔比星(Acla)等蒽环类药物联合组成HAD、HAA等方案,可进一步提高CR率。剂量增加的诱导化疗能提高1疗程CR率和缓解质量,但治疗相关毒性亦随之增加。中、大剂量Ara-c联合蒽环类的方案不能提高CR率,但可延长年轻病人的DFS。1疗程获CR者DFS长,2个标准疗程仍未CR者提示存在原发耐药,需换化疗方案或行allo-HSCT。②APL:多采用全反式维A酸(ATRA)+蒽环类药物。ATRA作用于RARA可诱导带有PML-RARA的APL细胞分化成熟,剂量为20~45mg/(m³·d)。砷剂作用于PML,小剂量能诱导APL细胞分化,大剂量能诱导其凋亡。ATRA+蒽环类的基础上加用砷剂(如三氧化二砷,ATO)能缩短达GR时间。低/中危组和不能耐受蒽环类药物者采用ATRA+ATO双诱导。治疗过程中需警惕出现分化综合征(differential syndrome),初诊时白细胞计数较高及治疗后迅速上升者易发生,其机制可能与细胞因子大量释放和黏附分子表达增加有关。临床表现为发热、肌肉骨骼疼痛、呼吸窘迫、肺间质浸润、胸腔积液、心包积液、体重增加、低血压、急性肾衰竭甚至死亡。一旦出现上述任一表现,应给子糖皮质激素治疗,并予吸氧、利尿,可暂停ATRA。除分化综合征外,ATRA的其他不良反应有头痛、颅内压增高、肝功能损害等;ATO的其他不良反应有肝功能损害、心电图QT间期延长等。APL合并凝血功能障碍和出血者积极输注血小板、新鲜冷冻血浆和冷沉淀,可减少由出血导致的早期死亡。
缓解后治疗:①AML的CNSL发生率不到3%,对初诊WBC>40×10°/L、伴髓外病变、M,/M,、伴t(8;21)或inv(16)的病人应在CR后做脑脊液检查并鞘内预防性用药至少1次,以进行CNSL筛查。而APL病人CR后至少预防性鞘内用药3次。②AML(非APL)比ALL治疗时间明显缩短。③APL在获得分子学缓解后可采用化疗、ATRA以及砷剂等药物交替维持治疗近2年,期间应定期监测并维持PML-RARA融合基因阴性。
年龄小于60岁的AML病人,根据表6-9-3的危险度分组选择相应的治疗方案。预后不良组首选allo-HSCT;预后良好组(非APL)首选大剂量Ara-C为基础的化疗,复发后再行allo-HSCT;预后中等组,配型相合的allo-HSCT和大剂量Ara-C为主的化疗均可采用。无法行allo-HSCT的预后不良组、部分预后良好组以及预后中等组病人均可考虑行自体HSCT。无法进行危险度分组者参照预后中等组治疗,若初诊时白细胞≥100×10°/L,则按预后不良组治疗。因年龄、并发症等原因无法采用上述治疗者,也可用常规剂量的不同药物组成化疗方案轮换巩固维持,但仅10%~15%的病人能长期生存。
HD Ara-C的最严重并发症是小脑共济失调,发生后必须停药。皮疹、发热、眼结膜炎也常见,可用糖皮质激素常规预防。
复发和难治AML的治疗可选用以下方案:①无交义耐药的新药组成联合化疗方案;②中、大剂量阿糖胞苷组成的联合方案;③HSCT;④临床试验;如耐药逆转剂、新的靶向药物(如FLT3抑制剂等)、生物治疗等。再诱导达CR后应尽快行allo-HSCT。复发的APL选用ATO±ATRA再诱导,CR后融合基因转阴者行自体HSCT或砷剂(不适合移植者)巩固治疗,融合基因仍阳性者考虑allo-HSCT或临床试验。
老年急性白血病(AL)的治疗
多数大于60岁的AL病人化疗需减量用药,以降低治疗相关死亡率。少数体质好、支持条件佳者可采用类似年轻病人的方案治疗,有HLA相合同胞供体者可行减低剂量预处理的alla-HSCT。由MDS转化面来、继发于某些理化因素、耐药、重要器官功能不全、不良核型及基因突变携带者,更应强调个体化治疗,如采用表观遗传学调控药物治疗或支持治疗等。
慢性白血病
慢性髓系白血病(CML)治疗
慢性髓系白血病 CP期治疗
CML治疗应着重于慢性期早期,避免疾病转化,其治疗如下:
需合用羟基探和别嘌醇。对于白细胞计数极高或有白细胞淤滞症表现的CP病人,可以行治疗性白细胞单采。明确诊断后,首选伊马替尼。
第一代酪氨酸激酶抑制剂(TKI)甲磺酸伊马替尼(imatinib meaylate,IM)为2-苯胺嘧啶衍生物,能特异性阻断ATP在ABL激酶上的结合位置,使酪氨酸残基不能磷酸化,从而抑制BCR-ABL阳性细跑的增殖。IM治疗CML病人完全细胞遗传学缓解率92%,10年总体生存率(overall surrival,0S)可达84%。IM耐药与基因点突变、BCRABL基因扩增和表达增加、P糖蛋白过度表达有关,随意减停药物容易产生BCR-ABL激酶区的突变,发生继发性耐药。第二代TKI如尼洛替尼(nilotinib)、达沙簪尼(dasatinib)治疗CML能够获得更快、更深的分子学反应,逐渐成为CML一线治疗方案的可选药物。TKI治疗期间可发生白细胞、血小板减少和贫血的血液学毒性以及水肿、头痛、皮疹、胆红素升高等非血液学毒性。在开始TKI治疗后的第3个月,6个月,12个月,18个月进行疗效监测,对判定为治疗失败的病人需进行ABL激酶区基因突变检查,并根据突变形式以及病人对药物的反应更换TKI或考虑造血于细胞移植。服药的依从性以及严密监测对于获得最佳疗效非常关键。
干扰素(intereron-a,IFN-a)是分子靶向药物出现之前的首选药物。目的用于不适合TKI和allo-HSCT的病人 。常用剂量300万 ~ 500万U/(m² ·d), 皮下或肌内注射,每周3 - 7次,坚持使用,推荐和小剂量阿糖胞苷(cytarabine,Ara-C) 合用 ,Ara-C常用剂量10~20mg/(m² ·d),每个月连用10天CCYR率约13%,有效者10年生存率可达70%,约50%的有效者可以长期生存。主要副作用包括乏力、发热、头痛、食欲缺乏、肌肉骨骼酸痛等流感样症状和体重下降、肝功能异常等,可引起轻到中度的血细胞减少。预防性使用对乙酰氨基酚等能够减轻流感样症状。
羟基脲(hydroxyurea,HU)细胞周期特异性化疗药,起效快,用药后两三天白细胞计数即下降,停药后又很快回升。常用剂量为3g/d,分2次口服,待白细胞减至20×10/L左右时,剂量减半。降至10×10'/L时,改为小剂量(0.5-1g/d)维持治疗。需经常检查血象,以便调节药物剂量。耐受性好,单独应用HU的CP病人中位生存期约为5年。单独应用HU目前限于高龄、具有合并症、TKI和IFN-a均不耐受的病人以及用于高白细胞淤滞时的降白细胞处理。其他药物包括Ara-C、高三尖杉酯碱(homoharningtonine,HHT)、砷剂、白消安等。
Allo-HSCT是CML的根治性治疗方法,但在CML慢性期不作为一线选择。Allo-HSCT仅用于移植风险很低且对TKI耐药、不耐受以及进展期的CML病人。
慢性髓系白血病进展期(AP和BC)的治疗
AP和BC统称为CML的进展期。CML进入进展期之后,需要评估病人的细胞遗传学、分子学BCR-ABL水平以及BCR-ABL激酶区的突变。AP病人,如果既往未使用过TKI治疗,可以采用加量的一代或者二代TKI(甲磺酸伊马替尼600-800mg/d或尼洛替尼800mg/d或达沙替尼140mg/d)使病人回到CP,立即行allo-HSCT治疗。BC病人,明确急变类型后,可以在加量的TKI基础上,加以联合化疗方案使病人回到CP后,立即行allo-HSCT治疗。Allo-HSCT干细胞来源不再受限于全相合供体,可以考虑行单倍型相合亲缘供体移植。移植后需辅以TKI治疗以减少复发,并可以行预防性供体淋巴细胞输注以增加移植物抗白血病效应。移植后复发可以用供体淋巴细胞输注联合或不联合TKI治疗以求再缓解。
进展期CML总体预后不住,明显不如CP的移植效果,TKI可以改善移植预后。有报道TK1联合allo-HSCT治疗进展期CML,3年OS达59%。
除allo HSCT外,进展期CML还可采用单用TK1,联合化疗,干扰素治疗或其他治疗,疗效有限且不能持久。
慢性淋巴细胞白血病(CLL)治疗
CLL为惰性白血病,并非所有病人在确诊后都需要立刻治疗。回顾性研究结果表明过早治疗并不能延长病人生存期,目前认为早期(Rai 0~Ⅱ期或Binet A期)病人无需治疗,定期随访即可。出现下列情况之一说明疾病处于活动状态,建议开始治疗:①疾病相关症状,包括6个月内无其他原因出现体重减少>10%、极度疲劳、非感染性发热(超过38℃)≥2周、盗汗;②巨脾(肋下缘>10cm)或进行性脾大及脾区疼痛;③淋巴结进行性肿大或直径>10cm;④进行性外周血淋巴细胞增多,2个月内增加>50%,或信增时间<6个月;⑤出现自身免疫性血细胞减少,糖皮质激素治疗无效;⑥骨简进行性衰竭,贫血和(或)血小板减少进行性加重。
既往CLL治疗多为姑息性,以减轻肿瘤负荷,改善症状为主要目的。近来新药物治疗效果不断提升,治疗后获得完全缓解(CR)的病人生存期较部分缓解和无效者延长。
烷化剂苯丁酸氮芥(chlorambucll,CLB),对初治CLL单药治疗反应率50%-60%,但CR率不足10%。目前多用于年龄较大、不能耐受其他药物化疗或有并发症的病人。环确酰胺的疗效与CLB相当,组成COP或CHOP方案并不优于单药。苯达莫司汀(bendamustme)是一种新型烷化剂,兼具有抗代谢功能和烷化剂作用,单药治疗CLL,不论是初治或复发难治性病人,均显示了较高的治疗反应率和CR率。
照呤类似物,氟达拉滨(fhudaabine,Fhu),总反应率60%-80%,CR率达20%~30%,中位缓解期约是CLB的2倍,但二者总生存期无差异。烷化剂耐药者换用Flu仍有效。嘌呤类似物联合烷化剂,如Fu联合环磷酰胺(FC方案),优于单用Hu,能有效延长初治CLL的无进展生存期,也可用于治疗难治性复发CLL。克拉屈滨、喷司他丁也可用于CLL的治疗,疗效、副作用与氟达拉滨相近。
.胰皮质激素,主要用于合并自身免疫性血细胞减少时的治疗,一般不单独应用,但大剂量甲泼尼龙对难治性CLL,尤其是17p缺失病人有较高的治疗反应率。
利妥昔单抗(rituximab)是人鼠嵌合型抗CD20单克隆抗体,对于表达CD20的CLL细胞有显著的治疗作用,但因CLL细胞表面CD20表达较少、血浆中存在可溶性CD20分子,利妥昔单抗在CLL病人体内清除过快,需加大剂量或密度才能有效。与阿仑单抗相比,利妥昔单抗潜在的免疫抑制作用。
利妥昔单抗联合化疗药物可以产生协同抗肿瘤效应,提高病人治疗的总体反应率和生存率。FC联合利妥昔单抗(FCR方案)治疗初治CLL,CR率可高达70%,总治疗反应率>90%,40%以上CR病人经PCR检测未发现徽小残留病灶。
CLL细胞内存在BTK、PBK、Syk等多种分子信号通路异常激活,针对以上信号通路的特异性抑制剂可能成为治疗CLL的药物。目前针对BTK通路的特异性抑制剂伊布替尼已经应用于CLL病人的一线和挽救治疗,单药伊布替尼一线治疗CLL的反应率达到90%,11%的病人达到CR,并且副作用较少。
大多数CLL病人无需一线接受造血干细胞移植,但是高危或复发难治病人可作为二线治疗。Allo-HSCT可使部分病人长期存恬甚至治意。但常规移植的相关并发症多,非清髓性移植(NST)可降低CLL移植相关死亡率,延长生存期。
因低γ球蛋白血症、中性粒细胞缺乏及老龄,CLI.病人极易感染,甚至导致病人死亡,因此应积极治疗和预防。反复感染或严重低γ球蛋白血症病人可静脉输注免疫球蛋白。并发AIHA或IIP者可用糖皮质激素治疗。有明显淋巴结肿大或巨脾、局部压迫症状明显者,在化疗效果不理想时,也可考虑放射治疗缓解症状。
预后
急性白血病
AL若不经治疗,平均生存期为3个月左右,短者甚至在确诊数天后死亡。经过现代治疗,不少病人可长期存活。对于ALL,1-9岁且白细胞<50×10'/L并伴有超二倍体或t(12;21)者预后最好,80%以上病人能够获得长期DFS甚至治愈。APL若能避免早期死亡则预后良好,多可治愈。老年、高白细胞的AL预后不良。继发性AL复发、多药耐药、需多疗程化疗方能缓解以及合并髓外白血病的AL预后较差。需要指出的是,某些指标的预后意义随治疗方法的改进而变化,如L,型B-ALL的预后经高剂量、短疗程、充分CNSL防治的强化治疗已大为改观,50%~60%的成人网人可以长期存活,加用抗CD20单克隆抗体后生存率进一步提高。
慢性白血病
慢性髓系白血病(CML)预后
TKI出现前,CML CP病人中位生存期为39-47个月,3~5年内进入BC终末期,少数病人CP可延续10~20年。影响CML预后的因素包括:病人初诊时的风险评估;疾病治疗的方式;病情的演变。干扰素治疗的OS较化疗有所提高,对干抗素的反应对预后有预示作用。TKI应用以来,生存期显著廷长。
慢性淋巴细胞白血病(CLL)预后
CLL是一种高度异质性疾病,从终身无需治疗到疾病短期快速进展,病程长短不一。CIL病人多死于骨髓衰竭导致的严重感染、贫血和出血。CLL.临床尚可发生转化,如Richer综合征、PUL等。近年来CLL的治疗发展迅速,单克隆抗体联合化疗的免疫化学治疗模式显著提高了病人的治疗反应率和生存率,而针对B细胞信号通路的特异性抑制剂等新型药物有望进一步提高临床疗效。
预防
高危人群
筛查建议
高危人群 :临床体检,每年 1 次。
临床体检包括以下项目 :
预防建议
历史
治疗史
1845年,白血病被称为全身性疾病;100多年后,1948年第一份关于白血病疗法的报告发布。直到1900年,白血病才被称为多因素疾病,而不是单一疾病。白血病的治疗探索经历了漫长的过程。
1865年,砷首次被利绍尔用于治疗白血病。砷是第一种在治疗某些形式的白血病方面具有一定益处的药物。伦琴于1895年发现X射线,这为白血病治疗带来了新的希望。1903年,美国外科医生尼古拉斯·森(英文:Nicholas Senn)首次将X射线疗法应用于白血病治疗。1942年,吉尔曼和飞利浦撰写了第一份关于氮芥对淋巴组织作用的临床报告,但由于战争形势,他们直到4年后才发表这一结果。1946年,多位学者发布研究结果表明氮芥对慢性白血病有积极作用。
西德尼·法伯(Sidney Farber)观察并认为,ALL等快速生长的肿瘤是由叶酸诱发的。因此,他推测叶酸拮抗剂可以抑制肿瘤生长。后来他报告了许多患有 ALL 的儿童暂时缓解的情况,但没有一个真正治愈。1943年,叶酸被认为是造血的重要物质。1946年,subbaRow首次将叶酸用于患者。1947年Sidney Farber与SubbaRow合作表明,叶酸拮抗剂可以对儿童急性白血病的治疗产生有效的影响。结果表明,儿童的改善是暂时的,但不是永久性的。1947年和1948年对16名患有急性白血病的婴儿和儿童进行的临床试验中,采用肌内注射的方式给予氨基蝶呤;这项干预措施使10名患者的临床、血液学和病理学证据得到重大改善。
西德尼·法伯于1948年提出现代白血病疗法,引入酸性叶酸作为抗癌疗法。法伯发表后,新的抗白血病药物问世。其中最著名的是嘌呤拮抗剂6-巯基嘌呤,1953年Burchenal和Lois Murphy证明它对儿童白血病有效。后来,其他抗白血病药物被引入,如1961年的L-天冬酰胺酶、1962年的长春新碱。关于新药疗法治疗过程的研究表明,自1964年至2014年,中位生存时间和长期生存明显改善。
发现史
白血病的早期历史可以追溯到200多年前。1811年,出生于格拉斯哥的爱丁堡医学教授彼得·卡伦用无法解释的乳白色血液定义了一例脾炎。威尔普于1825年定义了白血病的相关症。阿尔弗雷德·唐内在1844年发现了白细胞的成熟停滞。1845年约翰·贝内特根据显微镜下脓性白细胞的积累,将这种病命名为白血病。
研究史
阿尔弗雷德·唐内,于1844年,在其手稿中记录了对一名44岁患有脾肿大的女性的血液样本的观察结果。手稿中描述了从患者身上提取的血液的组织学形态,唐内注意到增加的原始细胞数量看起来与正常白细胞相同,并由此推断白细胞的增加是由分化停滞引起的,这目前仍然是白血病的主要特征。唐内是第一位对观察到的白血病患者的血液成分改变进行精确显微镜检查、描述和说明的医生。
英国医生约翰·贝内特于1845年将这一疾病命名为“白细胞增多症”,并首次指出白细胞的异常累积是一种全身性的血液疾病。同年,德国医生鲁道夫·魏尔啸在对一名50岁的女性进行的尸检中有了重要发现,他观察到红细胞数量减少,而白细胞数量增加。魏尔啸认为,血液外观的改变是由于正常生成的红细胞和白细胞之间的异常平衡,1845年11月,魏尔啸发表了关于“weisses blut”(德语,意为“白血”)的手稿。1847年,魏尔啸在检查了几个白血病病例后,第一个成功地区分了脾白血病和淋巴白血病。
1846年,时任圣乔治医院研究员的亨利·富勒(英文:Henry Fuller)发表了一篇病例报告,报告描述的患者脾脏和肝脏肿大,身体所有血管扩张,血液状况改变。富勒在患者生前进行了三次血液检查,患者去世后又检查了一次。在这些检查中,富勒发现了许多异常白细胞,它们大多数都比正常细胞大很多(即白血病原始细胞),且至少占患者血液的25%。富勒是第一位诊断出患者生前患有白血病的医生。
流行病学
发病率
来自法国国际癌症研究机构的研究人员根据184个国家白血病的流行病学模式,估算了全球白血病发病负担,该研究结果发表于2018年1月份的《柳叶刀》的血液学期刊。2003年至2007年间有717863例病例被纳入该分析,2012年估计有超过35万例新的白血病病例。
世界地区之间和内部的发病率存在很大差异。据统计,男女白血病发病率较高的是澳大利亚、新西兰、北美和西欧,最低的是西非。男性的患病比率通常高于女性,总体男女比例为1.4:1。在儿童中,ALL是所有研究国家中男女的主要亚型。在成人中的亚型分布要更加多样化,在大多数的欧洲和北美国家,CLL的比例相对更高,而在南美、加勒比海、亚洲和非洲的某些群体中,成人中ALL的发病率相对较高。
中国白血病发病率为(3〜4)/10万,在恶性肿瘤所致的死亡率中,白血病在男性中居第6位,在女性中居第7位,在儿童及35岁以下成人中,则居第1位。中国AL比CL多见(约5.5:1),其中AML最((1.62/10万),其次为ALL(0. 69/10万),CML(0. 39/10万),CLL少见(0. 05/10万)。男性发病率略高于女性(1.81:1)。成人AL中以AML多见,儿童以ALL多见,CML随年龄增长而发病率逐渐升高,CLL在50岁以后发病才明显增多。中国白血病发病率与亚洲其他国家相近,低于欧美国家。尤其是CLL不足白血病总发病率的5%,而在欧美国家则占25%-30%。
死亡率
白血病是一种高死亡率的疾病。世界范围内死亡率最高的是西欧、大洋洲、北美洲;男性患者的年死亡率为4.8/10万~7.4/10万,女性患者为 3.2/10万~4.6/10万。亚洲和拉丁美洲患者死亡率较低(男性患者为3.7/10万~4.5/10万,女性患者为2.8/10万~3.5/10万)。
自1950年以后,美国白人白血病患者的死亡率略有下降,非白人患者则稍有增加。自20世纪60年代早期开始,白人和非白人儿童和青少年(0~ 19岁)患者的死亡率显著下降,而年轻成人(20~44岁)患者的死亡率下降则相对缓慢;1950~ 1996年间中年人患者的死亡率无明显变化。中国1973~1975年白血病死亡率:男性患者为2.8/10万,女性患者为2.2/10万;1981~1982年天津市白血病死亡率为3.55/10万。
自20世纪90年代开始,美国、欧洲的急性髓系白血病的死亡率均呈下降趋势。急性髓系白血病的死亡率与年龄、性别、种族也有一定关系,随年龄的增长,死亡率增高(到80岁以上死亡率达高峰,17.6/10万)。2000~2003年间,美国急性髓系白血病的死亡率为2.7/10万;男性患者为3.5/10万,女性患者为2.2/10万。2000年美国白人急性髓系白血病的死亡率为2.7/10万,黑人为2.2/10万。
公共卫生
2022年12月,在中国关于政协第十三届全国委员会第五次会议第01046号(医疗卫生类102号)提案答复的函中,中华人民共和国国家卫生健康委员会表示将加快儿童恶性肿瘤优质医疗资源布局、加大儿童恶性肿瘤的医疗保障力度、加强儿童健康管理早期筛查力度并提高儿童肿瘤治疗的专业性。在儿童恶性肿瘤用药保障方面,治疗儿童费城染色体阳性的慢性髓性白血病药物尼洛替尼、儿童急性淋巴细胞白血病患者的一线治疗药物培门冬酶注射液等已纳入医保药品目录。
相关人物
鲁道夫·魏尔啸
鲁道夫·魏尔啸(德语:Rudolf Virchow,1821年10月13日-1902年9月5日),德国医生、人类学家、病理学家、史前史学家、生物学家、作家、编辑和政治家,以促进公共卫生而闻名,被誉为“现代病理学之父”。1845年,魏尔啸在对一名50岁的女性进行的尸检中有了重要发现,这一患者因腿部水肿、脾脏肿大、腹泻和鼻出血等症状而住进了夏里特医院。她的病情迅速恶化,并在三个月内死亡。魏尔啸在显微镜下发现了脓样物质,他观察到红细胞数量减少,而白细胞数量增加。魏尔啸认为,血液外观的改变是由于正常生成的红细胞和白细胞之间的异常平衡,而这种逆转的平衡似乎抑制了红细胞。1845年11月,魏尔啸发表了关于“weisses blut”(德语,意为“白血”)的手稿。
彼得·卡伦
彼得·卡伦(英文:Peter Cullen),爱丁堡医学教授,出生在格拉斯哥,于1786年完成了温盖特博士的学习生涯,1792年通过考试后成为一名外科医生。1810年6月14日,卡伦检查了一名35岁的男性患者,患者出现了明显的腹痛和发热。卡伦在其血液样本中观察到,血清在颜色和稠度上都像牛奶一样。在此之前,卡伦从未观察到过这种“乳白色”的血清。卡伦认为,这是由于脂肪的快速吸收从而形成的天然乳液。他的同事胡珀博士检查了病人的血液样本,注意到血清几乎完全由可凝固的淋巴液组成。这一发现与卡伦的想法不一致,卡伦的观点并不能解释乳白色血清与患者脾脏急性增生之间的联系。1811年,卡伦用无法解释的乳白色血液定义了这一例“脾炎”。根据目前历史资料,卡伦很可能是第一个描述慢性白血病病例的人。
阿尔弗雷德·维尔波
阿尔弗雷德·维尔波(法语:Alfred Velpeau,1795年-1867年),法国医生,于1823年获得医学博士学位,于1833年成为巴黎大学外科系主任。1827年,维尔波在巴黎皇家医学院发表了一份详细的尸检报告,病例为一位63岁的男性患者,该患者有明显的腹部肿胀、发热、无力和尿路结石等症状。其症状始于54岁,该疾病的稳定阶段持续了三年,之后患者因炎症而出现反复发烧,并陆续出现排尿困难、肿瘤占位等情况。维尔波在进行尸检的过程中,其巨大的肝脏和脾脏立即引起了他的注意。患者的血液成分也发生了显著的改变,如同稀粥一样稠厚,看起来就像充满脓液的血液。维尔波在报告中描述了这些相关症状,这些症状现在仍运用于白血病的早期诊断。维尔波描述了白血病症状与白细胞增多(牛乳状血清)之间的联系,并怀疑这种特殊的疾病与循环系统有关。
阿尔弗雷德·弗朗索瓦·唐内
阿尔弗雷德·弗朗索瓦·唐内(法语:Alfred François Donné,1801-1878),显微镜、微生物学和血液学的先驱,阴道毛滴虫的发现者。
1839年,一名44岁患有脾肿大的女性在巴黎迪厄医院去世,唐内检查了该患者的血液,观察到血液中一半的微粒看起来像“粘液小球”。1844年,唐内在其手稿中简要记录了他的观察结果。手稿中描述了从患者身上提取的血液的组织学形态,唐内认为,医生将血液中的“粘液小球”(即白细胞)的积聚误认为是脓液。他注意到增加的原始细胞数量看起来与正常白细胞相同,这一发现让唐内认为,白细胞的增加是由分化停滞引起的,这目前仍然是白血病的主要特征。遗憾的是,唐内没有描述他检查的白血病患者的相关症状,这名患者很可能患有慢性髓细胞性白血病。唐内是第一位对观察到的白血病患者的血液成分改变进行精确显微镜检查、描述和说明的医生。
约翰·贝内特
约翰·贝内特(英文:John Bennett),1833年在爱丁堡开始了他的医学研究,并于1837年毕业。同年,获得了“大脑生理学和病理学”的金牌。1841年教授组织学,强调显微镜对研究疾病的重要性,于1843年被任命为爱丁堡学院教授。
1845年,贝内特检查了一名28岁脾脏和肝脏肥大的男性患者的尸体,尸检显示直肠外侧上痔静脉有肿瘤,全身淋巴结肿大,并在血管中发现浓稠的奶油状脓液。显微镜检查显示,患者的血清由凝固的纤维蛋白丝组成,与许多无色小体(即白细胞)混合。贝内特描述了这一疾病的症状,并将其命名为“白细胞增多症”,被称为感染引起的疾病的继发性表现。贝内特是第一位指出白细胞的异常累积是一种全身性的血液疾病的医生。
亨利·富勒
亨利·富勒(英文:Henry Fuller),曼彻斯特圣乔治医院医生及医学法学讲师。富勒于1846年当选为圣乔治医院的研究员,1862年成为理事会成员,1868年成为副主席。
1846年,富勒发表了关于一名22岁的男性患者的病例报告,该患者脾脏和肝脏肿大,身体所有血管扩张,血液状况改变。其主要症状是四肢疼痛、头痛、头晕、直立性眩晕、无法入睡、食欲不振、恶心、呕吐等。尸检显示一个巨大的肿瘤占据了他的左腹部。富勒在患者生前进行了三次血液检查,患者去世后又检查了一次。在这些检查中,富勒发现了许多异常白细胞,它们大多数都比正常细胞大很多(即白血病原始细胞),且至少占患者血液的25%。富勒的病例报告准确地描述了异常白细胞的组成,是最早诊断出患者生前患有白血病的医生。
附表
附表1:AL的FAB分型
类型 | 描述 |
AML分型 |
M0急性髓细胞白血病微分化型 | 骨髓原始细胞>30% |
M1急性粒细胞白血病未分化型 | 原粒细胞(Ⅰ型+Ⅱ型,原粒细胞质中无颗粒为Ⅰ型,出现少数颗粒为Ⅱ型)占骨髓非红系有核细胞(NEC,指不包括浆细胞、淋巴细胞、组织嗜碱细胞、巨噬细胞及所有红系有核细胞的骨髓有核细胞计数)的90%以上 |
M2急性粒细胞白血病部分分化型 | 原粒细胞占骨髓NEC的30%~89%,其他粒细胞≥10%,单核细胞<20% |
M3急性早幼粒细胞白血病 (acute promyelocytic leukemia,APL) | 骨髓中以颗粒增多的早幼粒细胞为主,此类细胞在NEC中≥30% |
M4急性粒-单核细胞白血病 | 骨髓中原始细胞占NEC的30%以上,各阶段粒细胞≥20%,各阶段单核细胞≥20%; M4Eo:除上述M4型特点外,嗜酸性粒细胞在NEC中≥5% |
M5急性单核细胞白血病 | 骨髓NEC中原单核、幼单核≥30%,且原单核、幼单核及单核细胞≥80%,如果原单核细胞≥80%即为M5a,<80%为M5b |
M6红白血病 | 骨髓中幼红细胞≥50%,NEC中原始细胞(Ⅰ型+Ⅱ型)≥30% |
M7急性巨核细胞白血病 | 骨髓中原始巨核细胞≥30%,血小板抗原阳性,血小板过氧化酶阳性 |
ALL分型 |
L1 | 原始和幼淋巴细胞以小细胞(直径≤12µm)为主 |
L2 | 原始和幼淋巴细胞以大细胞(直径>12µm)为主 |
L3(Burkitt型) | 原始和幼淋巴细胞以大细胞为主,大小较一致,细胞内有明显空泡,胞质嗜碱性,染色深 |
附表2:AL的WHO分型
分型 | 描述(染色体异常) |
AML分型 |
伴重现性遗传学异常的AML | AML伴t(8;21)(q22;q22.1);RUNXI-RUNX1T1 AML伴inv(16)(p13.1q22)或t(16;16)(p13.1;q22);CBFB-MYH11 APL伴PML-RARA AML伴t(9;11)(p21.3;q23.3);MLLT3-KMT2A AML伴t(6;9)(p23;q34.1);DEK-NUP214 AML伴inv(3)(q21.3;q26.2)或t(3;3)(q21.3;q26.2);GATA2,MECOM AML(原始巨核细胞性)伴t(1;22)(p13.3;q13.3);RBM15-MKL1 AML伴BCR-ABL1 AML伴NPM1突变 AML伴CEBPA双等位基因突变 RUNX1突变 |
|
|
|
|
|
|
|
|
|
|
AML伴骨髓增生异常相关改变 | — |
治疗相关AML | — |
非特殊类型AML | AML微分化型 AML未分化型 AML部分分化型 急性粒-单核细胞白血病 急性单核细胞白血病 纯红血病 急性巨核细胞白血病 急性嗜碱性粒细胞白血病 急性全髓增生伴骨髓纤维化 |
髓系肉瘤 | — |
Down综合征相关的髓系增殖 | 短暂性异常骨髓增殖 Down综合征相关的髓系白血病 |
ALL分型 |
原始B淋巴细胞白血病 | B-ALL,非特指型 伴重现性遗传学异常的B-ALL B-ALL,BCR-ABL1样 B-ALL伴21号染色体内部扩增(iAMP21) |
原始T淋巴细胞白血病 | 早期前体T淋巴细胞白血病(ETP-ALL) 自然杀伤(NK)细胞白血病 |